GENOMIC
Mapping
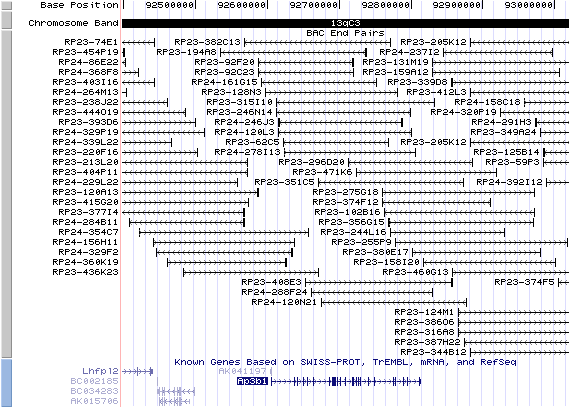
13qC3. View the map and BAC contig (data from UCSC genome browser).
Structure
(assembly 10/03)
Ap3b1/NM_009680: 27 exons, 207,285bp, chr13:92,605,147-92,812,431.
Note: Alternate name is adaptor-related protein complex 3 beta 1 (Ap3b1 or A3b1).
The figure below shows the structure of the Ap3b1 gene (data from UCSC genome browser).

Regulatory Element
Search the 5'UTR and 1kb upstream regions (human and mouse) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
Ap3b1/NM_009680: 3,956bp, view ORF and the alignment to genomic.
Expression Pattern
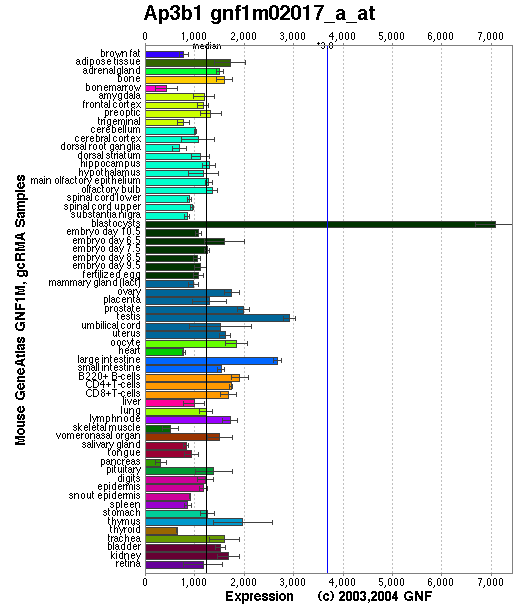
Tissue specificity: ubiquitously expressed. Ap3b1 produces a mRNA transcript of ~4.2-kb, detectable by Northern blots in kidney, heart, bone marrow, eye, and macrophages.
Affymetrix microarray expression pattern in SymAtlas from GNF is shown below.
PROTEIN
Sequence
Beta3A-adaptin (NP_033810): 1105aa, ExPaSy NiceProt view of Swiss-Prot:Q9Z1T1.
Synonyms: Adapter-related protein complex 3 beta 1 subunit; Adaptor protein complex AP-3 beta-1 subunit; AP-3 complex beta-1 subunit; Clathrin assembly protein complex 3 beta-1 large chain.
Ortholog
Species
Human Dog Rat Fruitfly Yeast GeneView
HPS2/AP3B1 AY221640 Ap3b1 CG11427 (rb) APL6 Protein
NP_033810 (1105aa) AAP45786 (1091aa) XM_226666 (1171aa) AAF71924 (1160aa) Apl6p (809aa) Identities
86% /965aa 85% /951aa 86% /1028aa 49% /575 26% /209
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc.
Domain
(1) Domains predicted by SMART:
a) low complexity: 10 - 24
b) low complexity: 275 - 291
c) coiled coil: 677 - 700
d) low complexity: 761 - 802
(2) Transmembrane domains predicted by SOSUI: none.
(3) Pfam domains: PF01602 - Adaptin N terminal region.
(4) CDD conserved domain: KOG1060, vesicle coat complex AP-3, beta subunit (Intracellular trafficking, secretion, and vesicular transport).
(5) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a) N-glycosylation site [pattern] [Warning: pattern with a high probability of occurrence]:
75 - 78 NASE, 402 - 405 NIST, 861 - 864 NLST, 870 - 873 NVST, 919 - 922 NTSD, 983 - 986 NVTL, 1015 - 1018 NETS.
b) Tyrosine sulfation site [rule] [Warning: rule with a high probability of occurrence]:
267 - 281 edneknfYeseeeee.
c) cAMP- and cGMP-dependent protein kinase phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
288 - 291 RKkS, 752 - 755 KRnS, 758 - 761 KRkS.
d) Tyrosine kinase phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
345 - 352 RsnrEvq.Y
e) Neutral zinc metallopeptidases, zinc-binding region signature [pattern]:
879 - 888 TktHELLHrM.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: found KLPI at 923
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: too long tail
i) Dileucine motif in the tail: found LL at 114
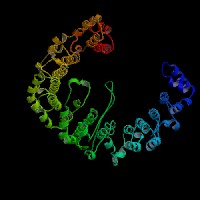
3D Model
ModBase: predicted comparative 3D structures on Q9Z1T1 (data from UCSC Gene Sorter). (from left to right: Front, Top, Side view)
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=122,870Da, pI=5.49 (NP_033810).
FUNCTION
Ontology
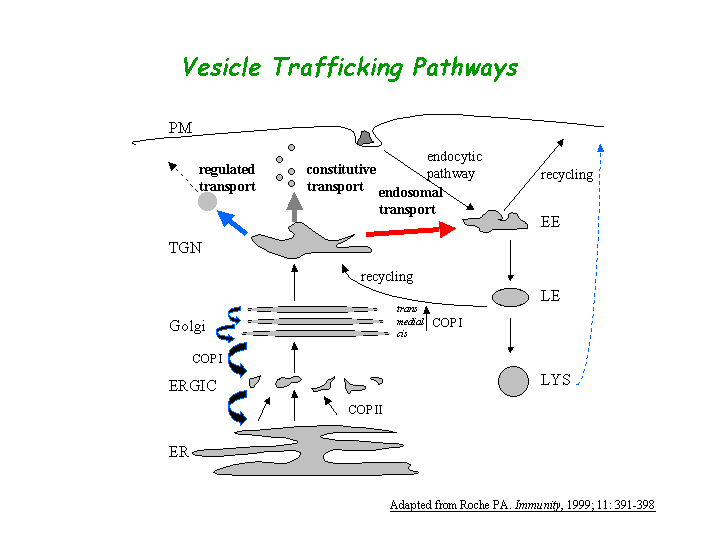
a) Biological process: intracellular protein transport (overview of trafficking pathway here).
b) Biological process: endocytsis
c) Component of Golgi apparatus.
d) Plays a role in protein sorting in the late-Golgi/trans-Golgi network (TGN) and/or endosomes.
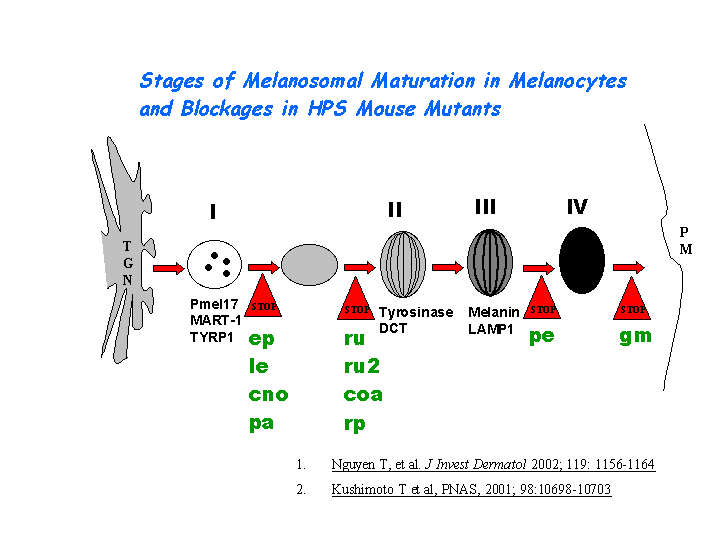
e) Plays a role in the vesicular trafficking of tyrosinase to melanosomes (view diagram of melanosomal protein sorting here).
Location
Component of the coat surrounding the cytoplasmic face of coated vesicles located at the Golgi complex.
Interaction
The Ap3b1 gene encodes the adapter-related protein complex 3 beta 1 subunit (beta-adaptin 3A, or AP-3 complex beta-3A subunit).
The AP-3 complex is a heterotetramer composed of two large adaptins (delta/AP3D1 and beta3A/AP3B2 or beta3B/AP3B1),
a medium adaptin (mu3A/AP3M1 or mu3B/AP3M2) and a small adaptin (sigma3A/AP3S1 or sigma3B/AP3S2).
In addition, the AP3M1 subunit interacts with tyrosinase for lysosomal targeting (Honing, et al).
In pearl cells, the delta and sigma3 subunits coassemble into a heterodimer, whereas mu3 gets destroyed.
The hinge and/or ear domains of beta3 are important for function, but the clathrin binding site is not needed
(Peden, et al). AP-3 complex interacts with CD1 antigen presenting molecules. Benson, et al
reported that C-terminal processing of neutrophil elastase exposes an AP3 interaction signal responsible for redirecting neutrophil elastase
trafficking from membranes to granules. More cargoes for AP-3 complex are reviewed by Dell'Angelica (2009).
6 proteins
are shown to be associated with APL6 in Yeast GRID.
Ap3b1 drosophila homolog
CG11427 interaction information
in CuraGen
interaction database.
Pathway
AP-3 complex is associated with the Golgi region as well as more peripheral structures. It facilitates the budding of vesicles from the Golgi membrane and appears to be involved in the sorting of a subset of transmembrane proteins targeted to lysosomes and lysosome-related organelles. In yeast, AP-3 sorts the vacuolar membrane enzymes,alkaline phosphatase and Vamp3p, a vacuolar t-SNARE (Odorizzi, et al). Faundez, et al found that the AP-3 complex is involved in synaptic vesicle formation in neuronal cells. Likewise, Sugita, et al proposed that there is an AP-3 dependent pathway for antigen presentation by CD1B (human) or CD1d (mice) molecules. More details in the description of the AP-3 dependent pathways are descriped in the human HPS2.
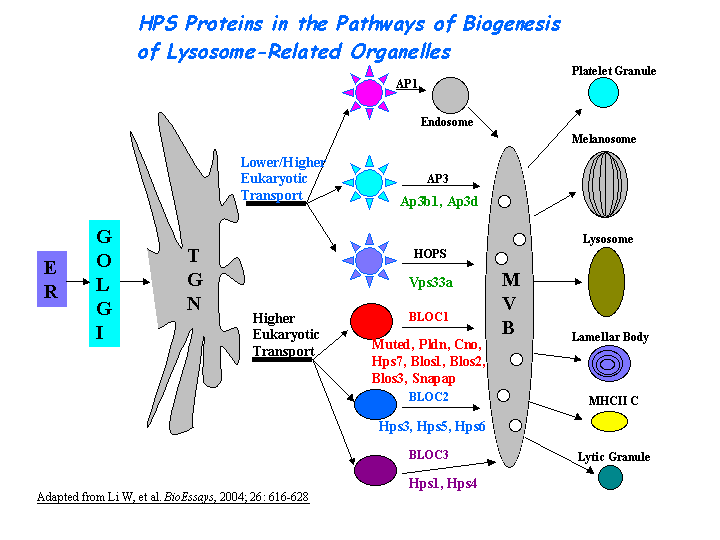
AP-3 interacts with HOPS complex or BLOC-1 in protein trafficking to lysosomes or LROs. However, the mechanism of AP-3 dependent pathway is distinct from that dependent on HPS1 (BLOC-3) (Feng, et al (2002))(view diagram of BLOC-3 and AP-3 pathway here). Hps2 may play a role in the late stages of the maturation of melanosomes (Nguyen, et al) (view diagram of melanosome blockage and melanosomal protein sorting here).
MUTATION
Allele or SNP
10 phenotypic alleles are described in MGI:1333879.
SNPs deposited in dbSNP.
Distribution
Location
Genomic
cDNA
Protein
Type
Strain
Reference
Exon 8
del exons 8~15
787G~1653G del 867bp (exons 8~15 )
D263~Q551 del 289aa
in-frame
pe-9J (B6)
Feng, et al (2000)
Exon 19
dup exons 19~24, ins 215bp partial transposon
2135A~2927G dup 793bp (exons 19~24)
E712dup 793bp
frame-shift, 976X
pe (B6)
Feng, et al (1999)
Exon 22
del exon 22
2504T~2610C del 107bp (exon 22)
W835del 107bp
frame-shift, 847X
pe-8J (DBA/2J)
Feng, et al (1999)
(Numbering of genomic and cDNA sequence is based on the start codon of RefSeq NM_009680.)
Effect
All the three allelic mutations involve in large deletion or insertion in the genome. The encoded Ap3b1 subunit or other subunits of AP3 complex are missing or destablized by Western blot analysis (Feng, et al (1999; 2000); Zhen, et al ).
PHENOTYPE
Defects in AP3B1 are the cause of the autosomal recessive phenotype 'pearl' (pe), a mouse model of Hermansky-Pudlak syndrome type 2 (HPS-2, OMIM 608233 ). The pe allele arose spontaneously from C3H/He, was transferred to C57BL/6J. The strain is described in more detail in JAX Mice database (B6.C3-Ap3b1pe/J). Homozygous mutants exhibit hypopigmentation, elevated kidney levels of lysosomal enzymes, platelet storage pool deficiency, reduced ipsilateral projections from the retina to brain (Novak, et al), reduced sensitivity of dark-adapted retina (night blindness) and shortened life span. In the Ap3b1/pe mutants, very small numbers of melanosomes are observed in the RPE, and melanosomes of the choroid are fewer in number and exhibit greater size heterogeneity than controls (Feng, et al (2002)). See more in details in Mouse Locus Card#Ap3b1. The transposonal mutation in pe may lead to the reversion of the mutation in peR (Feng, et al (2000)).
The CTL lytic granules present decreased secretion of in pearl mutant. CTL killing is severely impaired because of the loss of secretory lysosome polarization. The secretory lysosomes remain in the periphery of the cell, apparently stuck at the ends of microtubules (Clark, et al) (view diagram of lytic granule blockage in CTL cells here). AP-3 is most likely crucial for sorting a protein required for lysosomal polarization in CTL. Missorting of CD1d occurs in cells of the pearl and mocha (view diagram of CD1d blockage in APC cells here). In contrast, a wide variety of studies have noted normal peptide antigen presentation by the MHCII pathway in AP-3 deficient cells of mice and humans. Significantly lower NKT cell levels are found in spleen, thymus and liver of AP-3 deficient mice (Cernadas, et al). Benson, et al have found that frameshift mutations in the Ap3b1 subunit of AP-3 in the gray collie lead to unexpected consequences for levels of circulating leukocytes. The resulting loss of AP-3 function leads to hypopigmentation and the stem cell disease, cyclic hematopoiesis.
By homologous recombination, Yang, et al found that the Ap3b1 gene was disrupted in the null phenotype, Ap3b1(LN), which displayed phenotypes similar to those of pearl mice. Moreover, pearl is likely to be a hypomorph as the Ap3b1(LN) homozygotes had a lighter coat color and accumulated fewer of the mu3 and delta3 subunits of AP-3 than did pearl mice. Immunofluorescence analysis of fibroblasts and melanocytes cultured from Ap3b1(LN) homozygotes revealed that the lysosomal membrane proteins Lamp I and Lamp II and the melanosomal membrane protein tyrosinase were mislocalized. In particular, the Lamp proteins were clustered on the cell surface. These findings are similar to the observations in HPS-2 fibrablasts with the mislocalization of lysosomal proteins such as CD63, LAMP-1, LAMP-2, and LAMP-3 (Dell'Angelica, et al).
Lyerla et al demonstrated a mouse model of HPS, which is homozygously recessive for both the Hps1 (pale ear) and Hps2 (pearl) genes (Feng, et al (2002)), exhibits striking abnormalities of lung type II cells. Type II cells and lamellar bodies of this mutant are greatly enlarged, and the lamellar bodies are engorged with surfactant. Giant lamellar bodies (GLB) formation is not associated with abnormal trafficking or recycling of surfactant material. Instead, impaired secretion is an important component of GLB formation in ep/pe mice (Guttentag, et al). HPS double mutant ep/pe mouse strain develops interstitial pneumonia (HPSIP) past 1 year of age, which may be initiated by abnormal ATII cells and exacerbated by alveolar macrophage activation with elevated level of TGFbeta1 (Wang and Lyerla). Inflammation is initiated from the abnormal alveolar epithelial cells in ep/pe double mutant, and S-nitrosylated SP-D plays a significant role in amplifying pulmonary inflammation (Atochina-Vasserman, et al). Aberrant surfactant trafficking and secretion may lead to the apoptosis of alveolar epithelial type II cell in HPSIP, thereby causing the development of HPSIP (Mahavadi,et al). To study the pulmonary inflammation, cultured pearl alveolar macrophages (AMs) had markedly increased production of inflammatory cytokines at baseline, although baseline bronchoalveolar lavage (BAL) cell counts and differentials were similar in pearl and strain-matched wild-type mice. The lungs of ep and pe mice exhibit hyperresponsiveness to LPS and constitutive and organ-specific macrophage activation (Young, et al).
REFERENCE
- Atochina-Vasserman EN, Bates SR, Zhang P, Abramova H, Zhang Z, Gonzales L, Tao JQ, Gochuico BR, Gahl W, Guo CJ, Gow AJ, Beers MF, Guttentag S. Early alveolar epithelial dysfunction promotes lung inflammation in a mouse model of Hermansky-Pudlak syndrome. Am J Respir Crit Care Med 2011; 184: 449-58.PMID: 21616998
- Benson KF, Li FQ, Person RE, Albani D, Duan Z, Wechsler J, Meade-White K, Williams K, Acland GM, Niemeyer G, Lothrop CD, Horwitz M. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet 2003; 35: 90-6. PMID: 12897784
- Clark RH, Stinchcombe JC, Day A, Blott E, Booth S, Bossi G, Hamblin T, Davies EG, Griffiths GM. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat Immunol 2003; 4: 1111-20.
PMID:14566336
- Cernadas M, Sugita M, van der Wel N, Cao X, Gumperz JE, Maltsev S, Besra GS, Behar SM, Peters PJ, Brenner MB. Lysosomal localization of murine CD1d mediated by AP-3 is necessary for NK T cell development. J Immunol 2003; 171: 4149-55. PMID: 14530337
- Dell'Angelica EC. AP-3-dependent trafficking and disease: the first decade. Curr Opin Cell Biol 2009; 21: 552-9. Review. PMID: 19497727
- Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell 1999; 3: 11-21. PMID: 10024875
- Faundez V, Horng JT, Kelly RB. A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell 1998; 93: 423-32. PMID: 9590176
- Feng L, Rigatti BW, Novak EK, Gorin MB, Swank RT. Genomic structure of the mouse Ap3b1 gene in normal and pearl mice. Genomics 2000; 69: 370-9. PMID: 11056055
- Feng L, Novak EK, Hartnell LM, Bonifacino JS, Collinson LM, Swank RT. The Hermansky-Pudlak syndrome 1 (HPS1) and HPS2 genes independently contribute to the production and function of platelet dense granules, melanosomes, and lysosomes. Blood 2002; 99: 1651-8. PMID: 11861280
- Feng L, Seymour AB, Jiang S, To A, Peden AA, Novak EK, Zhen L, Rusiniak ME, Eicher EM, Robinson MS, Gorin MB, Swank RT. The beta3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum Mol Genet 1999; 8: 323-30. PMID: 9931340
- Guttentag SH, Akhtar A, Tao JQ, Atochina E, Rusiniak ME, Swank RT, Bates SR. Defective surfactant secretion in a mouse model of hermansky-pudlak syndrome. Am J Respir Cell Mol Biol 2005; 33:14-21. PMID: 1579097
- Honing S, Sandoval IV, von Figura K. A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosinase mediates selective binding of AP-3. EMBO J 1998; 17: 1304-14. PMID: 9482728
- Lyerla TA, Rusiniak ME, Borchers M, Jahreis G, Tan J, Ohtake P, Novak EK, Swank RT. Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am J Physiol Lung Cell Mol Physiol 2003; 285: L643-53. PMID: 12777251
- Mahavadi P, Korfei M, Henneke I, Liebisch G, Schmitz G, Gochuico BR, Markart P, Bellusci S, Seeger W, Ruppert C, Guenther A. Epithelial stress and apoptosis underlie Hermansky-Pudlak syndrome-associated interstitial pneumonia. Am J Respir Crit Care Med 2010;182: 207-19. PMID: 20378731
- Nguyen T, Novak EK, Kermani M, Fluhr J, Peters LL, Swank RT, Wei ML. Melanosome morphologies in murine models of hermansky-pudlak syndrome reflect blocks in organelle development. J Invest Dermatol 2002; 119: 1156-64. PMID: 12445206
- Novak EK, Hui SW, Swank RT. Platelet storage pool deficiency in mouse pigment mutations associated with seven distinct genetic loci. Blood 1984; 63: 536-44. PMID: 6696991
- Odorizzi G, Cowles CR, Emr SD. The AP-3 complex: a coat of many colours. Trends Cell Biol 1998; 8: 282-8. PMID: 9714600
- Peden AA, Rudge RE, Lui WW, Robinson MS. Assembly and function of AP-3 complexes in cells expressing mutant subunits. J Cell Biol 2002; 156: 327-36. PMID: 11807095
- Sugita M, Cao X, Watts GF, Rogers RA, Bonifacino JS, Brenner MB. Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity 2002; 16: 697-706. PMID: 12049721
- Wang L, Lyerla T. Histochemical and cellular changes accompanying the appearance of lung fibrosis in an experimental mouse model for Hermansky Pudlak syndrome. Histochem Cell Biol 2010; 134: 205-13. PMID: 20603711
- Yang W, Li C, Ward DM, Kaplan J, Mansour SL. Defective organellar membrane protein trafficking in Ap3b1-deficient cells. J Cell Sci 2000; 113: 4077-86. PMID: 11058094
- Young LR, Borchers MT, Allen HL, Gibbons RS, McCormack FX. Lung-restricted macrophage activation in the pearl mouse model of hermansky-pudlak syndrome. J Immunol 2006; 176: 4361-8. PMID: 16547274
- Zhen L, Jiang S, Feng L, Bright NA, Peden AA, Seymour AB, Novak EK, Elliott R, Gorin MB, Robinson MS, Swank RT. Abnormal expression and subcellular distribution of subunit proteins of the AP-3 adaptor complex lead to platelet storage pool deficiency in the pearl mouse. Blood 1999 ; 94: 146-55. PMID: 10381507
EDIT HISTORY:
Created by Wei Li: 06/21/2004
Updated by Wei Li: 04/05/2006
Updated by Wei Li: 07/29/2012
13qC3. View the map and BAC contig (data from UCSC genome browser).
Structure
(assembly 10/03)
Ap3b1/NM_009680: 27 exons, 207,285bp, chr13:92,605,147-92,812,431.
Note: Alternate name is adaptor-related protein complex 3 beta 1 (Ap3b1 or A3b1).
The figure below shows the structure of the Ap3b1 gene (data from UCSC genome browser).
Regulatory Element
Search the 5'UTR and 1kb upstream regions (human and mouse) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
Ap3b1/NM_009680: 3,956bp, view ORF and the alignment to genomic.
Expression Pattern
Tissue specificity: ubiquitously expressed. Ap3b1 produces a mRNA transcript of ~4.2-kb, detectable by Northern blots in kidney, heart, bone marrow, eye, and macrophages.
Affymetrix microarray expression pattern in SymAtlas from GNF is shown below.
PROTEIN
Sequence
Beta3A-adaptin (NP_033810): 1105aa, ExPaSy NiceProt view of Swiss-Prot:Q9Z1T1.
Synonyms: Adapter-related protein complex 3 beta 1 subunit; Adaptor protein complex AP-3 beta-1 subunit; AP-3 complex beta-1 subunit; Clathrin assembly protein complex 3 beta-1 large chain.
Ortholog
Species
Human Dog Rat Fruitfly Yeast GeneView
HPS2/AP3B1 AY221640 Ap3b1 CG11427 (rb) APL6 Protein
NP_033810 (1105aa) AAP45786 (1091aa) XM_226666 (1171aa) AAF71924 (1160aa) Apl6p (809aa) Identities
86% /965aa 85% /951aa 86% /1028aa 49% /575 26% /209
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc.
Domain
(1) Domains predicted by SMART:
a) low complexity: 10 - 24
b) low complexity: 275 - 291
c) coiled coil: 677 - 700
d) low complexity: 761 - 802
(2) Transmembrane domains predicted by SOSUI: none.
(3) Pfam domains: PF01602 - Adaptin N terminal region.
(4) CDD conserved domain: KOG1060, vesicle coat complex AP-3, beta subunit (Intracellular trafficking, secretion, and vesicular transport).
(5) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a) N-glycosylation site [pattern] [Warning: pattern with a high probability of occurrence]:
75 - 78 NASE, 402 - 405 NIST, 861 - 864 NLST, 870 - 873 NVST, 919 - 922 NTSD, 983 - 986 NVTL, 1015 - 1018 NETS.
b) Tyrosine sulfation site [rule] [Warning: rule with a high probability of occurrence]:
267 - 281 edneknfYeseeeee.
c) cAMP- and cGMP-dependent protein kinase phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
288 - 291 RKkS, 752 - 755 KRnS, 758 - 761 KRkS.
d) Tyrosine kinase phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
345 - 352 RsnrEvq.Y
e) Neutral zinc metallopeptidases, zinc-binding region signature [pattern]:
879 - 888 TktHELLHrM.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: found KLPI at 923
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: too long tail
i) Dileucine motif in the tail: found LL at 114
3D Model
ModBase: predicted comparative 3D structures on Q9Z1T1 (data from UCSC Gene Sorter). (from left to right: Front, Top, Side view)
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=122,870Da, pI=5.49 (NP_033810).
FUNCTION
Ontology
a) Biological process: intracellular protein transport (overview of trafficking pathway here).
b) Biological process: endocytsis
c) Component of Golgi apparatus.
d) Plays a role in protein sorting in the late-Golgi/trans-Golgi network (TGN) and/or endosomes.
e) Plays a role in the vesicular trafficking of tyrosinase to melanosomes (view diagram of melanosomal protein sorting here).
Location
Component of the coat surrounding the cytoplasmic face of coated vesicles located at the Golgi complex.
Interaction
The Ap3b1 gene encodes the adapter-related protein complex 3 beta 1 subunit (beta-adaptin 3A, or AP-3 complex beta-3A subunit).
The AP-3 complex is a heterotetramer composed of two large adaptins (delta/AP3D1 and beta3A/AP3B2 or beta3B/AP3B1),
a medium adaptin (mu3A/AP3M1 or mu3B/AP3M2) and a small adaptin (sigma3A/AP3S1 or sigma3B/AP3S2).
In addition, the AP3M1 subunit interacts with tyrosinase for lysosomal targeting (Honing, et al).
In pearl cells, the delta and sigma3 subunits coassemble into a heterodimer, whereas mu3 gets destroyed.
The hinge and/or ear domains of beta3 are important for function, but the clathrin binding site is not needed
(Peden, et al). AP-3 complex interacts with CD1 antigen presenting molecules. Benson, et al
reported that C-terminal processing of neutrophil elastase exposes an AP3 interaction signal responsible for redirecting neutrophil elastase
trafficking from membranes to granules. More cargoes for AP-3 complex are reviewed by Dell'Angelica (2009).
6 proteins
are shown to be associated with APL6 in Yeast GRID.
Ap3b1 drosophila homolog
CG11427 interaction information
in CuraGen
interaction database.
Pathway
AP-3 complex is associated with the Golgi region as well as more peripheral structures. It facilitates the budding of vesicles from the Golgi membrane and appears to be involved in the sorting of a subset of transmembrane proteins targeted to lysosomes and lysosome-related organelles. In yeast, AP-3 sorts the vacuolar membrane enzymes,alkaline phosphatase and Vamp3p, a vacuolar t-SNARE (Odorizzi, et al). Faundez, et al found that the AP-3 complex is involved in synaptic vesicle formation in neuronal cells. Likewise, Sugita, et al proposed that there is an AP-3 dependent pathway for antigen presentation by CD1B (human) or CD1d (mice) molecules. More details in the description of the AP-3 dependent pathways are descriped in the human HPS2.
AP-3 interacts with HOPS complex or BLOC-1 in protein trafficking to lysosomes or LROs. However, the mechanism of AP-3 dependent pathway is distinct from that dependent on HPS1 (BLOC-3) (Feng, et al (2002))(view diagram of BLOC-3 and AP-3 pathway here). Hps2 may play a role in the late stages of the maturation of melanosomes (Nguyen, et al) (view diagram of melanosome blockage and melanosomal protein sorting here).
MUTATION
Allele or SNP
10 phenotypic alleles are described in MGI:1333879.
SNPs deposited in dbSNP.
Distribution
Location
Genomic
cDNA
Protein
Type
Strain
Reference
Exon 8
del exons 8~15
787G~1653G del 867bp (exons 8~15 )
D263~Q551 del 289aa
in-frame
pe-9J (B6)
Feng, et al (2000)
Exon 19
dup exons 19~24, ins 215bp partial transposon
2135A~2927G dup 793bp (exons 19~24)
E712dup 793bp
frame-shift, 976X
pe (B6)
Feng, et al (1999)
Exon 22
del exon 22
2504T~2610C del 107bp (exon 22)
W835del 107bp
frame-shift, 847X
pe-8J (DBA/2J)
Feng, et al (1999)
(Numbering of genomic and cDNA sequence is based on the start codon of RefSeq NM_009680.)
Effect
All the three allelic mutations involve in large deletion or insertion in the genome. The encoded Ap3b1 subunit or other subunits of AP3 complex are missing or destablized by Western blot analysis (Feng, et al (1999; 2000); Zhen, et al ).
PHENOTYPE
Defects in AP3B1 are the cause of the autosomal recessive phenotype 'pearl' (pe), a mouse model of Hermansky-Pudlak syndrome type 2 (HPS-2, OMIM 608233 ). The pe allele arose spontaneously from C3H/He, was transferred to C57BL/6J. The strain is described in more detail in JAX Mice database (B6.C3-Ap3b1pe/J). Homozygous mutants exhibit hypopigmentation, elevated kidney levels of lysosomal enzymes, platelet storage pool deficiency, reduced ipsilateral projections from the retina to brain (Novak, et al), reduced sensitivity of dark-adapted retina (night blindness) and shortened life span. In the Ap3b1/pe mutants, very small numbers of melanosomes are observed in the RPE, and melanosomes of the choroid are fewer in number and exhibit greater size heterogeneity than controls (Feng, et al (2002)). See more in details in Mouse Locus Card#Ap3b1. The transposonal mutation in pe may lead to the reversion of the mutation in peR (Feng, et al (2000)).
The CTL lytic granules present decreased secretion of in pearl mutant. CTL killing is severely impaired because of the loss of secretory lysosome polarization. The secretory lysosomes remain in the periphery of the cell, apparently stuck at the ends of microtubules (Clark, et al) (view diagram of lytic granule blockage in CTL cells here). AP-3 is most likely crucial for sorting a protein required for lysosomal polarization in CTL. Missorting of CD1d occurs in cells of the pearl and mocha (view diagram of CD1d blockage in APC cells here). In contrast, a wide variety of studies have noted normal peptide antigen presentation by the MHCII pathway in AP-3 deficient cells of mice and humans. Significantly lower NKT cell levels are found in spleen, thymus and liver of AP-3 deficient mice (Cernadas, et al). Benson, et al have found that frameshift mutations in the Ap3b1 subunit of AP-3 in the gray collie lead to unexpected consequences for levels of circulating leukocytes. The resulting loss of AP-3 function leads to hypopigmentation and the stem cell disease, cyclic hematopoiesis.
By homologous recombination, Yang, et al found that the Ap3b1 gene was disrupted in the null phenotype, Ap3b1(LN), which displayed phenotypes similar to those of pearl mice. Moreover, pearl is likely to be a hypomorph as the Ap3b1(LN) homozygotes had a lighter coat color and accumulated fewer of the mu3 and delta3 subunits of AP-3 than did pearl mice. Immunofluorescence analysis of fibroblasts and melanocytes cultured from Ap3b1(LN) homozygotes revealed that the lysosomal membrane proteins Lamp I and Lamp II and the melanosomal membrane protein tyrosinase were mislocalized. In particular, the Lamp proteins were clustered on the cell surface. These findings are similar to the observations in HPS-2 fibrablasts with the mislocalization of lysosomal proteins such as CD63, LAMP-1, LAMP-2, and LAMP-3 (Dell'Angelica, et al).
Lyerla et al demonstrated a mouse model of HPS, which is homozygously recessive for both the Hps1 (pale ear) and Hps2 (pearl) genes (Feng, et al (2002)), exhibits striking abnormalities of lung type II cells. Type II cells and lamellar bodies of this mutant are greatly enlarged, and the lamellar bodies are engorged with surfactant. Giant lamellar bodies (GLB) formation is not associated with abnormal trafficking or recycling of surfactant material. Instead, impaired secretion is an important component of GLB formation in ep/pe mice (Guttentag, et al). HPS double mutant ep/pe mouse strain develops interstitial pneumonia (HPSIP) past 1 year of age, which may be initiated by abnormal ATII cells and exacerbated by alveolar macrophage activation with elevated level of TGFbeta1 (Wang and Lyerla). Inflammation is initiated from the abnormal alveolar epithelial cells in ep/pe double mutant, and S-nitrosylated SP-D plays a significant role in amplifying pulmonary inflammation (Atochina-Vasserman, et al). Aberrant surfactant trafficking and secretion may lead to the apoptosis of alveolar epithelial type II cell in HPSIP, thereby causing the development of HPSIP (Mahavadi,et al). To study the pulmonary inflammation, cultured pearl alveolar macrophages (AMs) had markedly increased production of inflammatory cytokines at baseline, although baseline bronchoalveolar lavage (BAL) cell counts and differentials were similar in pearl and strain-matched wild-type mice. The lungs of ep and pe mice exhibit hyperresponsiveness to LPS and constitutive and organ-specific macrophage activation (Young, et al).
REFERENCE
- Atochina-Vasserman EN, Bates SR, Zhang P, Abramova H, Zhang Z, Gonzales L, Tao JQ, Gochuico BR, Gahl W, Guo CJ, Gow AJ, Beers MF, Guttentag S. Early alveolar epithelial dysfunction promotes lung inflammation in a mouse model of Hermansky-Pudlak syndrome. Am J Respir Crit Care Med 2011; 184: 449-58.PMID: 21616998
- Benson KF, Li FQ, Person RE, Albani D, Duan Z, Wechsler J, Meade-White K, Williams K, Acland GM, Niemeyer G, Lothrop CD, Horwitz M. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet 2003; 35: 90-6. PMID: 12897784
- Clark RH, Stinchcombe JC, Day A, Blott E, Booth S, Bossi G, Hamblin T, Davies EG, Griffiths GM. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat Immunol 2003; 4: 1111-20.
PMID:14566336
- Cernadas M, Sugita M, van der Wel N, Cao X, Gumperz JE, Maltsev S, Besra GS, Behar SM, Peters PJ, Brenner MB. Lysosomal localization of murine CD1d mediated by AP-3 is necessary for NK T cell development. J Immunol 2003; 171: 4149-55. PMID: 14530337
- Dell'Angelica EC. AP-3-dependent trafficking and disease: the first decade. Curr Opin Cell Biol 2009; 21: 552-9. Review. PMID: 19497727
- Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell 1999; 3: 11-21. PMID: 10024875
- Faundez V, Horng JT, Kelly RB. A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell 1998; 93: 423-32. PMID: 9590176
- Feng L, Rigatti BW, Novak EK, Gorin MB, Swank RT. Genomic structure of the mouse Ap3b1 gene in normal and pearl mice. Genomics 2000; 69: 370-9. PMID: 11056055
- Feng L, Novak EK, Hartnell LM, Bonifacino JS, Collinson LM, Swank RT. The Hermansky-Pudlak syndrome 1 (HPS1) and HPS2 genes independently contribute to the production and function of platelet dense granules, melanosomes, and lysosomes. Blood 2002; 99: 1651-8. PMID: 11861280
- Feng L, Seymour AB, Jiang S, To A, Peden AA, Novak EK, Zhen L, Rusiniak ME, Eicher EM, Robinson MS, Gorin MB, Swank RT. The beta3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum Mol Genet 1999; 8: 323-30. PMID: 9931340
- Guttentag SH, Akhtar A, Tao JQ, Atochina E, Rusiniak ME, Swank RT, Bates SR. Defective surfactant secretion in a mouse model of hermansky-pudlak syndrome. Am J Respir Cell Mol Biol 2005; 33:14-21. PMID: 1579097
- Honing S, Sandoval IV, von Figura K. A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosinase mediates selective binding of AP-3. EMBO J 1998; 17: 1304-14. PMID: 9482728
- Lyerla TA, Rusiniak ME, Borchers M, Jahreis G, Tan J, Ohtake P, Novak EK, Swank RT. Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am J Physiol Lung Cell Mol Physiol 2003; 285: L643-53. PMID: 12777251
- Mahavadi P, Korfei M, Henneke I, Liebisch G, Schmitz G, Gochuico BR, Markart P, Bellusci S, Seeger W, Ruppert C, Guenther A. Epithelial stress and apoptosis underlie Hermansky-Pudlak syndrome-associated interstitial pneumonia. Am J Respir Crit Care Med 2010;182: 207-19. PMID: 20378731
- Nguyen T, Novak EK, Kermani M, Fluhr J, Peters LL, Swank RT, Wei ML. Melanosome morphologies in murine models of hermansky-pudlak syndrome reflect blocks in organelle development. J Invest Dermatol 2002; 119: 1156-64. PMID: 12445206
- Novak EK, Hui SW, Swank RT. Platelet storage pool deficiency in mouse pigment mutations associated with seven distinct genetic loci. Blood 1984; 63: 536-44. PMID: 6696991
- Odorizzi G, Cowles CR, Emr SD. The AP-3 complex: a coat of many colours. Trends Cell Biol 1998; 8: 282-8. PMID: 9714600
- Peden AA, Rudge RE, Lui WW, Robinson MS. Assembly and function of AP-3 complexes in cells expressing mutant subunits. J Cell Biol 2002; 156: 327-36. PMID: 11807095
- Sugita M, Cao X, Watts GF, Rogers RA, Bonifacino JS, Brenner MB. Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity 2002; 16: 697-706. PMID: 12049721
- Wang L, Lyerla T. Histochemical and cellular changes accompanying the appearance of lung fibrosis in an experimental mouse model for Hermansky Pudlak syndrome. Histochem Cell Biol 2010; 134: 205-13. PMID: 20603711
- Yang W, Li C, Ward DM, Kaplan J, Mansour SL. Defective organellar membrane protein trafficking in Ap3b1-deficient cells. J Cell Sci 2000; 113: 4077-86. PMID: 11058094
- Young LR, Borchers MT, Allen HL, Gibbons RS, McCormack FX. Lung-restricted macrophage activation in the pearl mouse model of hermansky-pudlak syndrome. J Immunol 2006; 176: 4361-8. PMID: 16547274
- Zhen L, Jiang S, Feng L, Bright NA, Peden AA, Seymour AB, Novak EK, Elliott R, Gorin MB, Robinson MS, Swank RT. Abnormal expression and subcellular distribution of subunit proteins of the AP-3 adaptor complex lead to platelet storage pool deficiency in the pearl mouse. Blood 1999 ; 94: 146-55. PMID: 10381507
EDIT HISTORY:
Created by Wei Li: 06/21/2004
Updated by Wei Li: 04/05/2006
Updated by Wei Li: 07/29/2012
(assembly 10/03)
Ap3b1/NM_009680: 27 exons, 207,285bp, chr13:92,605,147-92,812,431.
Note: Alternate name is adaptor-related protein complex 3 beta 1 (Ap3b1 or A3b1).
The figure below shows the structure of the Ap3b1 gene (data from UCSC genome browser).
Regulatory Element
Search the 5'UTR and 1kb upstream regions (human and mouse) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
Search the 5'UTR and 1kb upstream regions (human and mouse) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
Ap3b1/NM_009680: 3,956bp, view ORF and the alignment to genomic.
Expression Pattern
Tissue specificity: ubiquitously expressed. Ap3b1 produces a mRNA transcript of ~4.2-kb, detectable by Northern blots in kidney, heart, bone marrow, eye, and macrophages.
Affymetrix microarray expression pattern in SymAtlas from GNF is shown below.
Tissue specificity: ubiquitously expressed. Ap3b1 produces a mRNA transcript of ~4.2-kb, detectable by Northern blots in kidney, heart, bone marrow, eye, and macrophages.
Affymetrix microarray expression pattern in SymAtlas from GNF is shown below.
PROTEIN
Sequence
Beta3A-adaptin (NP_033810): 1105aa, ExPaSy NiceProt view of Swiss-Prot:Q9Z1T1.
Synonyms: Adapter-related protein complex 3 beta 1 subunit; Adaptor protein complex AP-3 beta-1 subunit; AP-3 complex beta-1 subunit; Clathrin assembly protein complex 3 beta-1 large chain.
Ortholog
| Species | Human | Dog | Rat | Fruitfly | Yeast |
| GeneView | HPS2/AP3B1 | AY221640 | Ap3b1 | CG11427 (rb) | APL6 |
| Protein | NP_033810 (1105aa) | AAP45786 (1091aa) | XM_226666 (1171aa) | AAF71924 (1160aa) | Apl6p (809aa) |
| Identities | 86% /965aa | 85% /951aa | 86% /1028aa | 49% /575 | 26% /209 |
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc.
Domain
(1) Domains predicted by SMART:
a) low complexity: 10 - 24
b) low complexity: 275 - 291
c) coiled coil: 677 - 700
d) low complexity: 761 - 802
(2) Transmembrane domains predicted by SOSUI: none.
(3) Pfam domains: PF01602 - Adaptin N terminal region.
(4) CDD conserved domain: KOG1060, vesicle coat complex AP-3, beta subunit (Intracellular trafficking, secretion, and vesicular transport).
(5) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a) N-glycosylation site [pattern] [Warning: pattern with a high probability of occurrence]:
75 - 78 NASE, 402 - 405 NIST, 861 - 864 NLST, 870 - 873 NVST, 919 - 922 NTSD, 983 - 986 NVTL, 1015 - 1018 NETS.
b) Tyrosine sulfation site [rule] [Warning: rule with a high probability of occurrence]:
267 - 281 edneknfYeseeeee.
c) cAMP- and cGMP-dependent protein kinase phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
288 - 291 RKkS, 752 - 755 KRnS, 758 - 761 KRkS.
d) Tyrosine kinase phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
345 - 352 RsnrEvq.Y
e) Neutral zinc metallopeptidases, zinc-binding region signature [pattern]:
879 - 888 TktHELLHrM.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: found KLPI at 923
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: too long tail
i) Dileucine motif in the tail: found LL at 114
3D Model
ModBase: predicted comparative 3D structures on Q9Z1T1 (data from UCSC Gene Sorter). (from left to right: Front, Top, Side view)
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=122,870Da, pI=5.49 (NP_033810).
(1) Domains predicted by SMART:
a) low complexity: 10 - 24
b) low complexity: 275 - 291
c) coiled coil: 677 - 700
d) low complexity: 761 - 802
(2) Transmembrane domains predicted by SOSUI: none.
(3) Pfam domains: PF01602 - Adaptin N terminal region.
(4) CDD conserved domain: KOG1060, vesicle coat complex AP-3, beta subunit (Intracellular trafficking, secretion, and vesicular transport).
(5) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a) N-glycosylation site [pattern] [Warning: pattern with a high probability of occurrence]:
75 - 78 NASE, 402 - 405 NIST, 861 - 864 NLST, 870 - 873 NVST, 919 - 922 NTSD, 983 - 986 NVTL, 1015 - 1018 NETS.
b) Tyrosine sulfation site [rule] [Warning: rule with a high probability of occurrence]:
267 - 281 edneknfYeseeeee.
c) cAMP- and cGMP-dependent protein kinase phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
288 - 291 RKkS, 752 - 755 KRnS, 758 - 761 KRkS.
d) Tyrosine kinase phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
345 - 352 RsnrEvq.Y
e) Neutral zinc metallopeptidases, zinc-binding region signature [pattern]:
879 - 888 TktHELLHrM.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: found KLPI at 923
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: too long tail
i) Dileucine motif in the tail: found LL at 114
3D Model
ModBase: predicted comparative 3D structures on Q9Z1T1 (data from UCSC Gene Sorter). (from left to right: Front, Top, Side view)
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=122,870Da, pI=5.49 (NP_033810).
FUNCTION
Ontology
a) Biological process: intracellular protein transport (overview of trafficking pathway here).
b) Biological process: endocytsis
c) Component of Golgi apparatus.
d) Plays a role in protein sorting in the late-Golgi/trans-Golgi network (TGN) and/or endosomes.
e) Plays a role in the vesicular trafficking of tyrosinase to melanosomes (view diagram of melanosomal protein sorting here).
{kind=link}
{kind=link}
Location
Component of the coat surrounding the cytoplasmic face of coated vesicles located at the Golgi complex.
Interaction
The Ap3b1 gene encodes the adapter-related protein complex 3 beta 1 subunit (beta-adaptin 3A, or AP-3 complex beta-3A subunit). The AP-3 complex is a heterotetramer composed of two large adaptins (delta/AP3D1 and beta3A/AP3B2 or beta3B/AP3B1), a medium adaptin (mu3A/AP3M1 or mu3B/AP3M2) and a small adaptin (sigma3A/AP3S1 or sigma3B/AP3S2). In addition, the AP3M1 subunit interacts with tyrosinase for lysosomal targeting (Honing, et al). In pearl cells, the delta and sigma3 subunits coassemble into a heterodimer, whereas mu3 gets destroyed. The hinge and/or ear domains of beta3 are important for function, but the clathrin binding site is not needed (Peden, et al). AP-3 complex interacts with CD1 antigen presenting molecules. Benson, et al reported that C-terminal processing of neutrophil elastase exposes an AP3 interaction signal responsible for redirecting neutrophil elastase trafficking from membranes to granules. More cargoes for AP-3 complex are reviewed by Dell'Angelica (2009).
6 proteins are shown to be associated with APL6 in Yeast GRID.
Ap3b1 drosophila homolog CG11427 interaction information in CuraGen interaction database.
Pathway
AP-3 complex is associated with the Golgi region as well as more peripheral structures. It facilitates the budding of vesicles from the Golgi membrane and appears to be involved in the sorting of a subset of transmembrane proteins targeted to lysosomes and lysosome-related organelles. In yeast, AP-3 sorts the vacuolar membrane enzymes,alkaline phosphatase and Vamp3p, a vacuolar t-SNARE (Odorizzi, et al). Faundez, et al found that the AP-3 complex is involved in synaptic vesicle formation in neuronal cells. Likewise, Sugita, et al proposed that there is an AP-3 dependent pathway for antigen presentation by CD1B (human) or CD1d (mice) molecules. More details in the description of the AP-3 dependent pathways are descriped in the human HPS2.
AP-3 interacts with HOPS complex or BLOC-1 in protein trafficking to lysosomes or LROs. However, the mechanism of AP-3 dependent pathway is distinct from that dependent on HPS1 (BLOC-3) (Feng, et al (2002))(view diagram of BLOC-3 and AP-3 pathway here). Hps2 may play a role in the late stages of the maturation of melanosomes (Nguyen, et al) (view diagram of melanosome blockage and melanosomal protein sorting here).
{kind=link}
MUTATION
Allele or SNP
10 phenotypic alleles are described in MGI:1333879.
SNPs deposited in dbSNP.
Distribution
| Location | Genomic | cDNA | Protein | Type | Strain | Reference |
| Exon 8 | del exons 8~15 | 787G~1653G del 867bp (exons 8~15 ) | D263~Q551 del 289aa | in-frame | pe-9J (B6) | Feng, et al (2000) |
| Exon 19 | dup exons 19~24, ins 215bp partial transposon | 2135A~2927G dup 793bp (exons 19~24) | E712dup 793bp | frame-shift, 976X | pe (B6) | Feng, et al (1999) |
| Exon 22 | del exon 22 | 2504T~2610C del 107bp (exon 22) | W835del 107bp | frame-shift, 847X | pe-8J (DBA/2J) | Feng, et al (1999) |
Effect
All the three allelic mutations involve in large deletion or insertion in the genome. The encoded Ap3b1 subunit or other subunits of AP3 complex are missing or destablized by Western blot analysis (Feng, et al (1999; 2000); Zhen, et al ).
PHENOTYPE
Defects in AP3B1 are the cause of the autosomal recessive phenotype 'pearl' (pe), a mouse model of Hermansky-Pudlak syndrome type 2 (HPS-2, OMIM 608233 ). The pe allele arose spontaneously from C3H/He, was transferred to C57BL/6J. The strain is described in more detail in JAX Mice database (B6.C3-Ap3b1pe/J). Homozygous mutants exhibit hypopigmentation, elevated kidney levels of lysosomal enzymes, platelet storage pool deficiency, reduced ipsilateral projections from the retina to brain (Novak, et al), reduced sensitivity of dark-adapted retina (night blindness) and shortened life span. In the Ap3b1/pe mutants, very small numbers of melanosomes are observed in the RPE, and melanosomes of the choroid are fewer in number and exhibit greater size heterogeneity than controls (Feng, et al (2002)). See more in details in Mouse Locus Card#Ap3b1. The transposonal mutation in pe may lead to the reversion of the mutation in peR (Feng, et al (2000)).
The CTL lytic granules present decreased secretion of in pearl mutant. CTL killing is severely impaired because of the loss of secretory lysosome polarization. The secretory lysosomes remain in the periphery of the cell, apparently stuck at the ends of microtubules (Clark, et al) (view diagram of lytic granule blockage in CTL cells here). AP-3 is most likely crucial for sorting a protein required for lysosomal polarization in CTL. Missorting of CD1d occurs in cells of the pearl and mocha (view diagram of CD1d blockage in APC cells here). In contrast, a wide variety of studies have noted normal peptide antigen presentation by the MHCII pathway in AP-3 deficient cells of mice and humans. Significantly lower NKT cell levels are found in spleen, thymus and liver of AP-3 deficient mice (Cernadas, et al). Benson, et al have found that frameshift mutations in the Ap3b1 subunit of AP-3 in the gray collie lead to unexpected consequences for levels of circulating leukocytes. The resulting loss of AP-3 function leads to hypopigmentation and the stem cell disease, cyclic hematopoiesis.
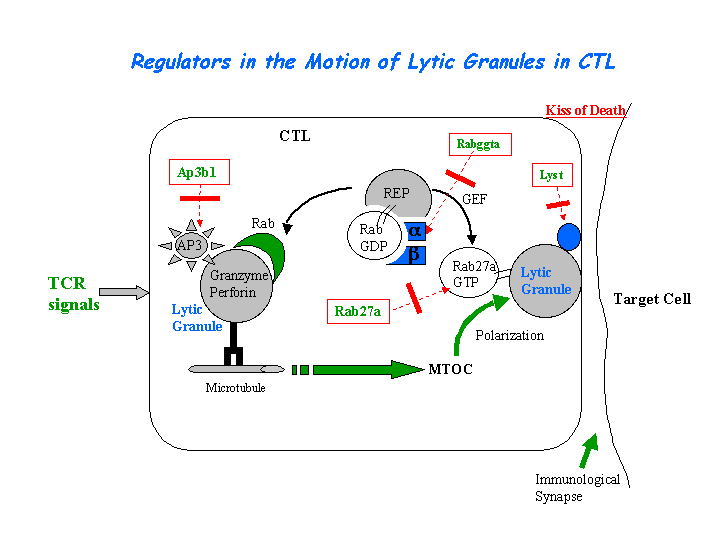{kind=link}
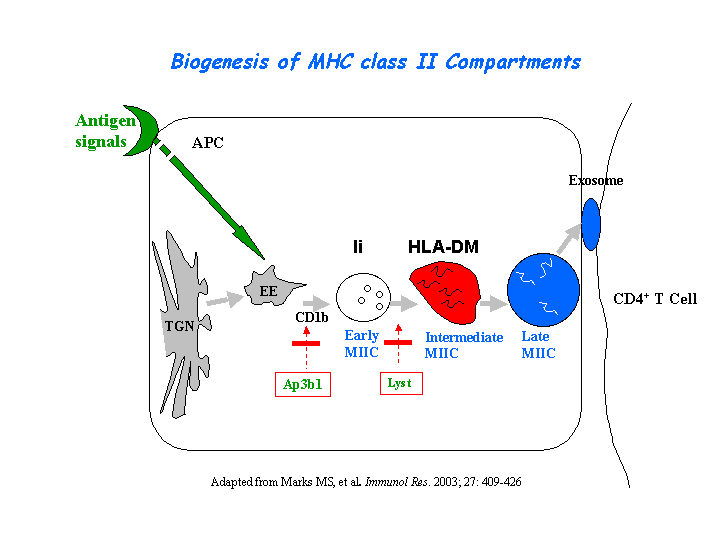{kind=link}
By homologous recombination, Yang, et al found that the Ap3b1 gene was disrupted in the null phenotype, Ap3b1(LN), which displayed phenotypes similar to those of pearl mice. Moreover, pearl is likely to be a hypomorph as the Ap3b1(LN) homozygotes had a lighter coat color and accumulated fewer of the mu3 and delta3 subunits of AP-3 than did pearl mice. Immunofluorescence analysis of fibroblasts and melanocytes cultured from Ap3b1(LN) homozygotes revealed that the lysosomal membrane proteins Lamp I and Lamp II and the melanosomal membrane protein tyrosinase were mislocalized. In particular, the Lamp proteins were clustered on the cell surface. These findings are similar to the observations in HPS-2 fibrablasts with the mislocalization of lysosomal proteins such as CD63, LAMP-1, LAMP-2, and LAMP-3 (Dell'Angelica, et al).
Lyerla et al demonstrated a mouse model of HPS, which is homozygously recessive for both the Hps1 (pale ear) and Hps2 (pearl) genes (Feng, et al (2002)), exhibits striking abnormalities of lung type II cells. Type II cells and lamellar bodies of this mutant are greatly enlarged, and the lamellar bodies are engorged with surfactant. Giant lamellar bodies (GLB) formation is not associated with abnormal trafficking or recycling of surfactant material. Instead, impaired secretion is an important component of GLB formation in ep/pe mice (Guttentag, et al). HPS double mutant ep/pe mouse strain develops interstitial pneumonia (HPSIP) past 1 year of age, which may be initiated by abnormal ATII cells and exacerbated by alveolar macrophage activation with elevated level of TGFbeta1 (Wang and Lyerla). Inflammation is initiated from the abnormal alveolar epithelial cells in ep/pe double mutant, and S-nitrosylated SP-D plays a significant role in amplifying pulmonary inflammation (Atochina-Vasserman, et al). Aberrant surfactant trafficking and secretion may lead to the apoptosis of alveolar epithelial type II cell in HPSIP, thereby causing the development of HPSIP (Mahavadi,et al). To study the pulmonary inflammation, cultured pearl alveolar macrophages (AMs) had markedly increased production of inflammatory cytokines at baseline, although baseline bronchoalveolar lavage (BAL) cell counts and differentials were similar in pearl and strain-matched wild-type mice. The lungs of ep and pe mice exhibit hyperresponsiveness to LPS and constitutive and organ-specific macrophage activation (Young, et al).
REFERENCE
- Atochina-Vasserman EN, Bates SR, Zhang P, Abramova H, Zhang Z, Gonzales L, Tao JQ, Gochuico BR, Gahl W, Guo CJ, Gow AJ, Beers MF, Guttentag S. Early alveolar epithelial dysfunction promotes lung inflammation in a mouse model of Hermansky-Pudlak syndrome. Am J Respir Crit Care Med 2011; 184: 449-58.PMID: 21616998
- Benson KF, Li FQ, Person RE, Albani D, Duan Z, Wechsler J, Meade-White K, Williams K, Acland GM, Niemeyer G, Lothrop CD, Horwitz M. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet 2003; 35: 90-6. PMID: 12897784
- Clark RH, Stinchcombe JC, Day A, Blott E, Booth S, Bossi G, Hamblin T, Davies EG, Griffiths GM. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat Immunol 2003; 4: 1111-20. PMID:14566336
- Cernadas M, Sugita M, van der Wel N, Cao X, Gumperz JE, Maltsev S, Besra GS, Behar SM, Peters PJ, Brenner MB. Lysosomal localization of murine CD1d mediated by AP-3 is necessary for NK T cell development. J Immunol 2003; 171: 4149-55. PMID: 14530337
- Dell'Angelica EC. AP-3-dependent trafficking and disease: the first decade. Curr Opin Cell Biol 2009; 21: 552-9. Review. PMID: 19497727
- Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell 1999; 3: 11-21. PMID: 10024875
- Faundez V, Horng JT, Kelly RB. A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell 1998; 93: 423-32. PMID: 9590176
- Feng L, Rigatti BW, Novak EK, Gorin MB, Swank RT. Genomic structure of the mouse Ap3b1 gene in normal and pearl mice. Genomics 2000; 69: 370-9. PMID: 11056055
- Feng L, Novak EK, Hartnell LM, Bonifacino JS, Collinson LM, Swank RT. The Hermansky-Pudlak syndrome 1 (HPS1) and HPS2 genes independently contribute to the production and function of platelet dense granules, melanosomes, and lysosomes. Blood 2002; 99: 1651-8. PMID: 11861280
- Feng L, Seymour AB, Jiang S, To A, Peden AA, Novak EK, Zhen L, Rusiniak ME, Eicher EM, Robinson MS, Gorin MB, Swank RT. The beta3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum Mol Genet 1999; 8: 323-30. PMID: 9931340
- Guttentag SH, Akhtar A, Tao JQ, Atochina E, Rusiniak ME, Swank RT, Bates SR. Defective surfactant secretion in a mouse model of hermansky-pudlak syndrome. Am J Respir Cell Mol Biol 2005; 33:14-21. PMID: 1579097
- Honing S, Sandoval IV, von Figura K. A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosinase mediates selective binding of AP-3. EMBO J 1998; 17: 1304-14. PMID: 9482728
- Lyerla TA, Rusiniak ME, Borchers M, Jahreis G, Tan J, Ohtake P, Novak EK, Swank RT. Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am J Physiol Lung Cell Mol Physiol 2003; 285: L643-53. PMID: 12777251
- Mahavadi P, Korfei M, Henneke I, Liebisch G, Schmitz G, Gochuico BR, Markart P, Bellusci S, Seeger W, Ruppert C, Guenther A. Epithelial stress and apoptosis underlie Hermansky-Pudlak syndrome-associated interstitial pneumonia. Am J Respir Crit Care Med 2010;182: 207-19. PMID: 20378731
- Nguyen T, Novak EK, Kermani M, Fluhr J, Peters LL, Swank RT, Wei ML. Melanosome morphologies in murine models of hermansky-pudlak syndrome reflect blocks in organelle development. J Invest Dermatol 2002; 119: 1156-64. PMID: 12445206
- Novak EK, Hui SW, Swank RT. Platelet storage pool deficiency in mouse pigment mutations associated with seven distinct genetic loci. Blood 1984; 63: 536-44. PMID: 6696991
- Odorizzi G, Cowles CR, Emr SD. The AP-3 complex: a coat of many colours. Trends Cell Biol 1998; 8: 282-8. PMID: 9714600
- Peden AA, Rudge RE, Lui WW, Robinson MS. Assembly and function of AP-3 complexes in cells expressing mutant subunits. J Cell Biol 2002; 156: 327-36. PMID: 11807095
- Sugita M, Cao X, Watts GF, Rogers RA, Bonifacino JS, Brenner MB. Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity 2002; 16: 697-706. PMID: 12049721
- Wang L, Lyerla T. Histochemical and cellular changes accompanying the appearance of lung fibrosis in an experimental mouse model for Hermansky Pudlak syndrome. Histochem Cell Biol 2010; 134: 205-13. PMID: 20603711
- Yang W, Li C, Ward DM, Kaplan J, Mansour SL. Defective organellar membrane protein trafficking in Ap3b1-deficient cells. J Cell Sci 2000; 113: 4077-86. PMID: 11058094
- Young LR, Borchers MT, Allen HL, Gibbons RS, McCormack FX. Lung-restricted macrophage activation in the pearl mouse model of hermansky-pudlak syndrome. J Immunol 2006; 176: 4361-8. PMID: 16547274
- Zhen L, Jiang S, Feng L, Bright NA, Peden AA, Seymour AB, Novak EK, Elliott R, Gorin MB, Robinson MS, Swank RT. Abnormal expression and subcellular distribution of subunit proteins of the AP-3 adaptor complex lead to platelet storage pool deficiency in the pearl mouse. Blood 1999 ; 94: 146-55. PMID: 10381507
EDIT HISTORY:
Created by Wei Li: 06/21/2004
Updated by Wei Li: 04/05/2006
Updated by Wei Li: 07/29/2012