GENOMIC
Mapping
1q42.3; View the map and BAC clones (data from UCSC genome browser).
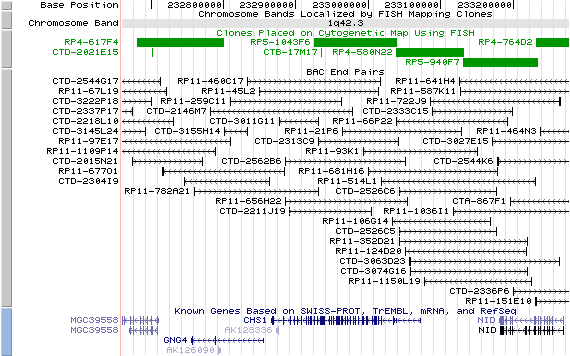
Structure
(assembly 07/03)
LYST/NM_000081: 54 exons, 205,873bp, Chr1: 232,865,497-233,071,369.
The figure below shows the structure of the LYST gene (data from UCSC genome browser).

Regulatory Element
Search the 5'UTR and 1kb upstream regions (human and mouse) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
LYST (NM_000081),
13,449bp, view ORF and the alignment to genomic.
Expression Pattern
Tissue specificity: ubiquitous. Abundantly expressed in adult and fetal thymus, peripheral blood leukocytes, bone marrow and several regions of the adult brain. On Northern blot analysis, the predominant LYST transcript is ~13.5kb. Minor 5.8kb transcript is arising from incomplete splicing and reading through an intron (Barbosa, et al (1997)). Additional alternative splice forms of LYST may exist.

BMR: Bone marrow; SPL: Spleen; TMS: Thymus; BRN: Brain; SPC: Spinal cord; HRT: Heart; MSL: Skeletal muscle;
LVR; Liver; PNC: Pancreas; PST: Prostate; KDN: Kidney; LNG: Lung. (data from GeneCards )
PROTEIN
Sequence
Lysosomal trafficking regulator (NP_000072): 3801aa, ExPaSy NiceProt view of Swiss-Prot:Q99698.
Synonyms: beige protein homolog.
Ortholog
| Species | Mouse | Rat | Zebrafish | HoneyBee | Fruitfly |
| GeneView | Lyst/bg | Lyst | 06386 | LOC408774 | CG11814 |
| Protein | NP_034878 (3788aa) | NP_445970 (3788aa) | 25336 (1292aa) | XP_392306 (3219aa) | NP_647681 (2517aa) |
| Identities | 84%/3228aa | 85%/3238aa | 50%/672aa | 27%/716aa | 26%/611aa |
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc.
Domain
(1) Domains predicted by SMART:
a) low complexity 22 - 36
b) low complexity 72 - 82
c) low complexity 399 - 412
d) low complexity 663 - 671
e) low complexity 1340 - 1351
f) low complexity 1373 - 1383
g) low complexity 2438 - 2456
h) low complexity 2545 - 2556
i) low complexity 2804 - 2817
j) Pfam:Beach: 3132 - 3422
k) Pfam:WD40: 3553 - 3593, 3605 - 3644, 3647 - 3690, 3738 - 3779
(2) Transmembrane domains predicted by SOSUI:
No. N terminal transmembrane region C terminal type length
1 2300 LVPICCGLYELLSGVLLILPDVL 2322 PRIMARY 23
2 2453 QILNSCSKVADMLLDNGLLYVLC 2475 SECONDARY 23
(3) Graphical view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a) BEACH domain profile :
3120 - 3422: score=137.273
b) Trp-Asp (WD) repeats profile :
3612 - 3646: score=10.575
c) Trp-Asp (WD) repeats circular profile :
3612 - 3788: score=17.113
d) Type-1 copper (blue) proteins signature :
1683 - 1697: Af.YlYaCg.PnHtsv.M
e) Trp-Asp (WD) repeats signature :
3631 - 3645: LISVsrDgtCiIWDL
f) cAMP- and cGMP-dependent protein kinase phosphorylation site : [occurs frequently]
147 - 150: RKiT,
885 - 888: RRkT,
1633 - 1636: RKtS,
2588 - 2591: KRkS,
2624 - 2627: RRmS.
g) Amidation site : [occurs frequently]
1009 - 1012: qGKK,
1493 - 1496: kGKK,
1604 - 1607: qGKR.
h) Tyrosine kinase phosphorylation site : [occurs frequently]
1213 - 1221: KllvEeegY,
1703 - 1711: KpvnDyskY,
3105 - 3111: Kvr.Ddv.Y,
3155 - 3163: RsfnDlmqY,
3198 - 3204: Kek.Edr.Y,
3203 - 3210: Ryv.DtykY,
3209 - 3215: Kyl.Eee.Y,
3342 - 3349: RqalEsd.Y.
i) Leucine zipper pattern : [occurs frequently]
2316 - 2337: LilpdvlLedvmdkLiqadtlL,
2371 - 2392: LknrgfsLlanqlyLhrgtqeL.
j) Bipartite nuclear targeting sequence : [occurs frequently]
144 - 160:
RRqrkithrysvrdark,
484 - 500:
KKvkseqlhhsmctrkr,
2145 - 2161:
KKqnslgssdtlkkgke.
k) Tyrosine sulfation site : [occurs frequently]
535 - 549:
etadgdvYyperccc,
536 - 550:
tadgdvyYperccci,
922 - 936:
dsedtsgYdstasep,
1214 - 1228:
llveeegYeadsesn,
1517 - 1531:
drpegaeYinpgerl,
3062 - 3076:
pasfswtYeeikevh.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: found TLPI at 780
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: none
i) Dileucine motif in the tail: none
3D Model
(1) ModBase matched entries found, results here.
(2)ModBase predicted comparative 3D structure of Q99698 from UCSC Genome Sorter.
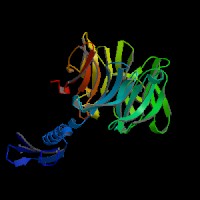
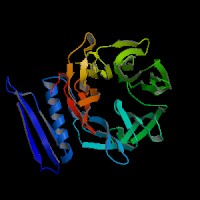
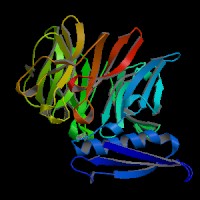
From left to right: Front, Top, and Side views of predicted protein.
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=429,125Da, pI=6.15 (NP_000072).
FUNCTION
Ontology
a) Biological process: intracellular protein transport from endosome to lysosome transport
b) Involved in cellular defense response
c) Component of endosome
d) Associated with microtubule cytoskeleton.
Location
Cytoplasmic.
Interaction
Twenty-one LYST-interacting proteins (LIPs) were identified in yeast two-hybrid screens by using fourteen cDNA fragments, covering the entire coding domain of LYST, as baits to screen five human cDNA libraries. Four confirmed interactions were with proteins important in vesicular transport and signal transduction (the SNARE-complex protein HRS, 14-3-3 tau, casein kinase II beta subunit, and troponin I) (Tchernev, et al). Fujita, et al identified the SBP1 by yeast two-hybrid screening, with the N-terminal half (1-157) of human SBP1 identical to lyst-interacting protein 5. SBP1 binds directly to SKD1, a member of the AAA-ATPase family and one of the mammalian class E Vps (vacuolar protein sorting) proteins, through its C-terminal region (198-309). They proposed that the SKD1-SBP1 complex, together with lyst protein, may function in endosomal membrane transport.
Lyst drosophila homolog CG11814 interaction information shown in CuraGen interaction database.
Pathway
The beige protein may be required for sorting endosomal resident proteins into late multivesicular endosomes by a mechanism involving microtubules (Faigle, et al).
MUTATION
Allele or SNP
26 mutations deposited in HGMD.
SNPs deposited in dbSNP.
10 selected allelic examples described in OMIM.
Distribution
The distribution of 14 mutations is described in detail in Albinism Database. Other 12 mutations are described in Karim et al. Two additional novel mutations on exon 5 and exon 43 were reported by Zarzour et al.
Effect
Most patients with CHS manifest with severe disease in childhood. Patients with severe childhood CHS have a functionally-null mutant CHS1 allele, whereas patients with the adolescent and adult forms of CHS have missense-mutant alleles that likely encode CHS1 polypeptides with partial function (Karim et al).
PHENOTYPE
Chediak-Higashi Syndrome (CHS, OMIM 214500) is a recessive autosomal disease caused by mutations in the CHS1/LYST gene (Barbosa, et al (1996)) (OMIM 606897). Patients with CHS exhibit hypopigmentation of the skin, eyes, and hair, prolonged bleeding times, easy bruisability, recurrent infections, abnormal natural killer cell function, and peripheral neuropathy. About 85 to 90% of CHS patients eventually develop a strange lymphoproliferative syndrome, the so-called 'accelerated phase' of the disorder, characterized by generalized lymphohistiocytic infiltrates, fever, jaundice, hepatosplenomegaly, lymphadenopathy, pancytopenia, and bleeding. Mortality results from frequent bacterial infections or from an "accelerated phase" lymphoproliferation into the major organs of the body. About 10-15 percent of patients exhibit milder clinical phenotypes and survive to adulthood, but develop progressive and often fatal neurological dysfunction with intellectual decline, tremor, ataxia, peripheral neuropathy, and white-matter deterioration, often resulting in death. The hallmark of CHS is giant inclusion bodies in all granulated cells, giant lysosomes, and giant melanosomes. The enlarged lysosomes seem to be the result of too much membrane fusion or insufficient membrane fission.
Peptide loading onto major histocompatibility complex class II molecules and antigen presentation are strongly delayed in the B cells from CHS patients. Only lysosomal multilaminar compartments are enlarged (reaching 1-2 micron), whereas late multivesicular endosomes have normal size and morphology. In contrast to giant multilaminar compartments that bear most of the usual lysosomal markers in these cells (HLA-DR, HLA-DM, Lamp-1, CD63, etc.), multivesicular late endosomes displayed reduced levels of all these molecules, suggesting a defect in transport from the trans-Golgi network and/or early endosomes into late multivesicular endosomes (Faigle, et al) (view diagram of blockage in APC cells here).
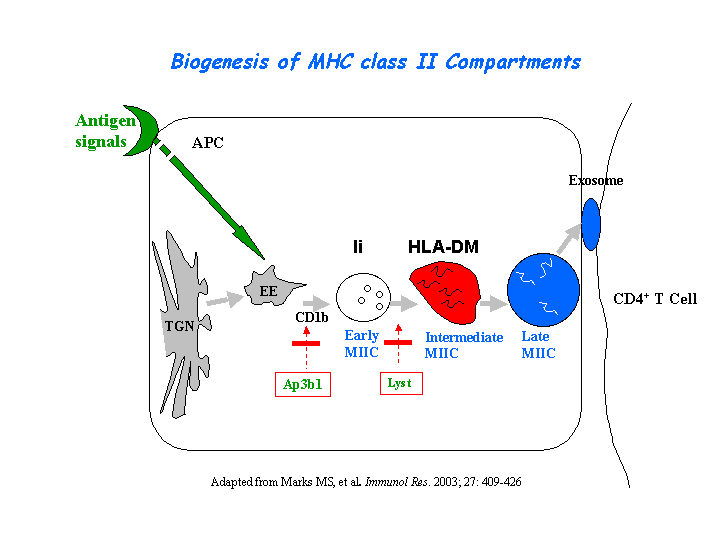{kind=link}
Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) is present in enlarged, abnormal vesicles in CHS T cells and is not properly expressed at the cell surface after T cell activation, whereas its surface expression is not impaired. The defective surface expression of CTLA-4 by CHS T cells may be involved in the generation of lymphoproliferative disease (Barrat, et al). The CTL defect arises from the loss of secretion at the immunological synapse. (view diagram of lytic granule blockage in CTL cells here).
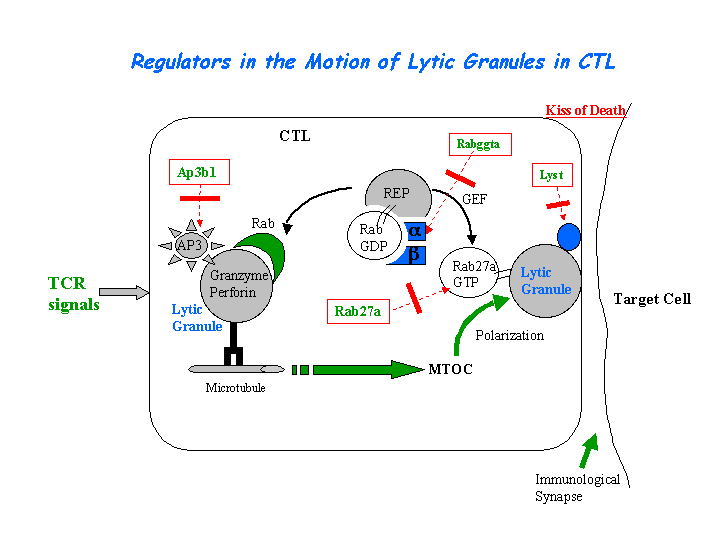{kind=link}
REFERENCE
- Barbosa MD, Barrat FJ, Tchernev VT, Nguyen QA, Mishra VS, Colman SD, Pastural E, Dufourcq-Lagelouse R, Fischer A, Holcombe RF, Wallace MR, Brandt SJ, de Saint Basile G, Kingsmore SF. Identification of mutations in two major mRNA isoforms of the Chediak-Higashi syndrome gene in human and mouse. Hum Mol Genet 1997; 6: 1091-8. PMID: 9215680
- Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, Brandt SJ, Chotai D, Hodgman C, Solari RC, Lovett M, Kingsmore SF. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 1996; 382: 262-5. PMID: 8717042
- Barrat FJ, Le Deist F, Benkerrou M, Bousso P, Feldmann J, Fischer A, de Saint Basile G. Defective CTLA-4 cycling pathway in Chediak-Higashi syndrome: a possible mechanism for deregulation of T lymphocyte activation. Proc Natl Acad Sci U S A 1999; 96: 8645-50. PMID: 10411929
- Faigle W, Raposo G, Tenza D, Pinet V, Vogt AB, Kropshofer H, Fischer A, de Saint-Basile G, Amigorena S. Deficient peptide loading and MHC class II endosomal sorting in a human genetic immunodeficiency disease: the Chediak-Higashi syndrome. J Cell Biol 1998; 141: 1121-34. PMID: 9606205
- Fujita H, Umezuki Y, Imamura K, Ishikawa D, Uchimura S, Nara A, Yoshimori T, Hayashizaki Y, Kawai J, Ishidoh K, Tanaka Y, Himeno M. Mammalian class E Vps proteins, SBP1 and mVps2/CHMP2A, interact with and regulate the function of an AAA-ATPase SKD1/Vps4B. J Cell Sci 2004; 117: 2997-3009. PMID: 15173323
- Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ, Barbosa E, Falik-Borenstein T, Filipovich A, Ishida Y, Kivrikko S, Klein C, Kreuz F, Levin A, Miyajima H, Regueiro J, Russo C, Uyama E, Vierimaa O, Spritz RA. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet 2002; 108: 16-22. PMID: 11857544
- Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, McGrail SH, Dussault BJ Jr, Perou CM, Boissy RE, Duyk GM, Spritz RA, Moore KJ. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet 1996; 14: 307-11. PMID: 8896560
- Tchernev VT, Mansfield TA, Giot L, Kumar AM, Nandabalan K, Li Y, Mishra VS, Detter JC, Rothberg JM, Wallace MR, Southwick FS, Kingsmore SF. The Chediak-Higashi protein interacts with SNARE complex and signal transduction proteins. Mol Med 2002; 8: 56-64. PMID: 11984006
- Zarzour W, Kleta R, Frangoul H, Suwannarat P, Jeong A, Kim SY, Wayne AS, Gunay-Aygun M, White J, Filipovich AH, Gahl WA. Two novel CHS1 (LYST) mutations: clinical correlations in an infant with Chediak-Higashi syndrome. Mol Genet Metab 2005; 85:125-32. PMID: 15896657
EDIT HISTORY:
Created by Wei Li & Jonathan Bourne: 07/13/2004
Updated by Wei Li: 12/28/2005