GENOMIC
Mapping
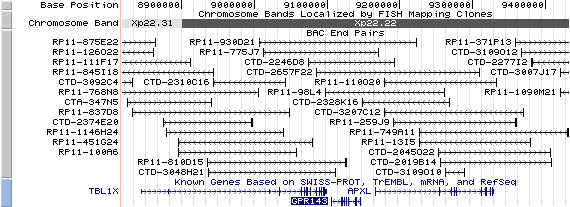
Xp22.22. View the map and BAC clones (data from UCSC genome browser).
Structure
(assembly 07/03)
GPR143/NM_000273: 9 exons, 40,464bp, ChrX: 9,104,959 - 9,145,422.
The figure below shows the structure of the GPR143 gene (data from UCSC genome browser).

Regulatory Element
Search the 5'UTR and 1kb upstream regions (human and mouse) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
GPR143 (NM_000273), 1,607bp, view ORF and the alignment to genomic.
Expression Pattern
Tissue specificity: Highly expressed in pigment cells. Maybe expressed in other tissues as shown below.
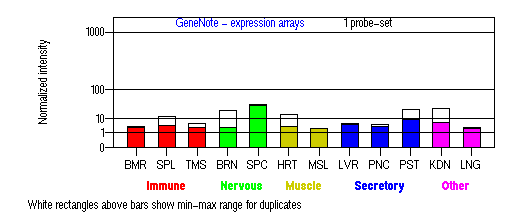
BMR: Bone marrow; SPL: Spleen; TMS: Thymus; BRN: Brain; SPC: Spinal cord; HRT: Heart; MSL: Skeletal muscle;
LVR; Liver; PNC: Pancreas; PST: Prostate; KDN: Kidney; LNG: Lung. (data from GeneCards )
PROTEIN
Sequence
G protein-coupled receptor 143 (NP_000264): 424aa, ExPaSy NiceProt view of Swiss-Prot:P51810.
Synonyms: Ocular albinism type 1 protein.
Ortholog
| Species | Mouse | Rat | Zebrafish | Mosquito | Fruitfly |
| GeneView | Gpr143 | LOC302619 | 393795 | 1277742 | CG4521/mthl1 |
| Protein | NP_035081 (405aa) | XP_228848 (464aa) | NP_957116 (412aa) | XP_317232 (321aa) | NP_573140 (676aa) |
| Identities | 77% /315aa | 75% /279aa | 60% /236aa | 31% /71aa | 19% /60aa |
View multiple sequence alignment (PDF files) by ClustalW and GeneDoc.
Domain
(1) Domains predicted by SMART:
a) low complexity: 5 - 18
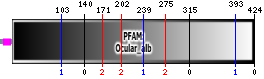
b) Pfam:Ocular_alb: 21 - 424
Pfam:Ocular_alb:
(2) Transmembrane domains predicted by SOSUI: six transmembrane helices detected.
| No. | N terminal | transmembrane region | C terminal | type | length |
| 1 | 90 | ILRAAAACDLLGCLGMVIRSTVW | 112 | SECONDARY | 23 |
| 2 | 136 | CVGSAMWIQLLYSACFWWLFCYA | 158 | PRIMARY | 23 |
| 3 | 172 | STILLYHIMAWGLATLLCVEGAA | 194 | PRIMARY | 23 |
| 4 | 210 | HAIPHYVTMYLPLLLVLVANPIL | 232 | SECONDARY | 23 |
| 5 | 265 | RFFKIMLVLIICWLSNIINESLL | 287 | PRIMARY | 23 |
| 6 | 309 | KTTWFIMGILNPAQGFLLSLAFY | 331 | SECONDARY | 23 |
Motif/Site
(1) Predicted results by ScanProsite:
a) Amidation site : [occurs frequently]
4 - 7: aGRR,
70 - 73: pGRR.
b) Protein kinase C phosphorylation site : [occurs frequently]
23 - 25: SpR,
87 - 89: SvR,
300 - 302: SlK,
342 - 344: SpR,
373 - 375: SgK.
c) N-myristoylation site : [occurs frequently]
58 - 63: GLrlAL,
78 - 83: GSpaTS,
207 - 212: GLdhAI,
335 - 340: GCslGF,
380 - 385: GGqtSD,
394 - 399: GSdaST.
d) Casein kinase II phosphorylation site : [occurs frequently]
121 - 124: SvsD,
202 - 205: SrcE,
252 - 255: TenE,
355 - 358: SaaE,
383 - 386: TsdE,
398 - 401: StiE,
404 - 407: TasE,
420 - 423: ThgD.
e) N-glycosylation site : [occurs frequently]
126 - 129: NHTE,
283 - 286: NESL.
Not that D106 is essential for N-glycosylation of the protein (Shen, et al (2001a)).
(2) Predicted results of subprograms by PSORT II:
a) Seems to no N-terminal signal peptide
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: none
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: none
i) Dileucine motif in the tail: none
3D Model
(1)No ModBase entry found.
(2) 3D models predicted by SPARKS (fold recognition) below. View the models by PDB2MGIF.

2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=46,051Da, pI=8.39 (NP_000264).
Two forms of the OA1 protein were identified by Western analysis, a 60-kDa glycoprotein and a doublet of 48 and 45 kDa probably corresponding to unglycosylated precursor polypeptides (Schiaffino, et al (1996)).
FUNCTION
Ontology
a) Biological process: eye pigment biosynthesis; visual perception
b) Biological process: G-protein coupled receptor protein signaling pathway
c) Integral to membrane
d) Belongs to family OA of G-protein coupled receptors.
Location
Late endo-lysosomal compartments (Shen, et al (2001b)).
Melanosomal membrane (Schiaffino, et al (1996; 1999)).
Interaction
OA1 protein is a conserved integral membrane protein with seven transmembrane domains that has weak similarities to G protein-coupled receptors (GPCRs), which participate in the most common signal transduction system at the plasma membrane. It binds heterotrimeric G proteins, which suggests that OA1 GPCR-mediated signal transduction systems also operate at the internal membranes in mammalian cells (Schiaffino, et al (1999) ). Two separate sorting signals that are both necessary and sufficient for intracellular retention, as well as lysosomal and melanosomal localization were identified. These sorting signals are an unconventional dileucine motif within the third cytosolic loop and a novel motif, characterized by a tryptophan-glutamic acid doublet, within the C-terminal tail (Piccirillo, et al). OA1 colocalizes and coprecipitates with arrestins, which downregulate the signaling of OA1 by specifically reducing its expression levels (Innamorati, et al). Oa1 is a target of MITF, and tissue-specific control of Oa1 transcription lies within a region of 617 bp that contains the E-box bound by Mitf (Vetrini, et al). L-DOPA is a ligand for OA1 (Lopez, et al).
The Drosophila homolog, CG4521, has no entries in the CuraGen Drosophila interaction database.
Pathway
The OA1 gene product is a membrane glycoprotein that may play a role in controlling melanosome growth and maturation. OA1 protein functions in the late stage of melanosome development.
MUTATION
Allele or SNP
78 mutations deposited in HGMD.
SNPs deposited in dbSNP.
Selected allelic examples described in OMIM.
Distribution
The distribution of 61 mutations and 3 polymorphisms is described in detail in Albinism Database. For cross-reference, view the distribution of mutations in the Retina International Mutation Database of OA1 Gene.
Except the 78 mutations deposited in HGMD, additional novel mutations are listed below:
(1) The S89F mutation was identified in a Chinese OA1 family by Liu, et al (2007).
(2) Eight novel mutations (W132R, E185K, R186P, R186W, c.616insC, c.737delG, c.780delC, and Y311X) were described by Roma, et al (2007).
(3) Two novel mutations, c.190delC and p.C116G, were reported in two United States OA1 family by Iannaccone, et al (2007).
(4) One novel mutation, c.222-258del37bp was reported in a Chinese OA1 family by Zhou, et al (2008), resulting in a truncated protein with 93 amino acids.
(5) Five novel mutations in GPR143 were identified in six Chinese OA1 families: c.849delT (p.Val284SerfsX15); c.238_240delCTC (p.Leu80del); c.658+1G>A, g.1103_7266del6164 (p.Gly84AlafsX65, deletion of exons 2 and 3); and g.25985_26546del562 (p.Gly296ValfsX26, deletion of exon 8) (Fang, et al (2008)).
(6) One novel nonsense mutation, G315X, was reported in a Chinese OA1 family by Wang, et al (2009).
(7) A previously unidentified 19 bp duplication (c.291_309) mutation in exon 1 was identified in a Chinese OA1 family by Peng, et al (2009).
(8)Micale, et al (2009) identified the c.564_565delCT and a novel intragenic deletion g.5360_6371del1012, encompassing exon 2, in two unrelated Italian families.
Effect
A number of mutations in the OA1 gene lead to at least in part the retention of the aberrant protein in the endoplasmic reticulum due to the protein misfolding (d'Addio, et al).
PHENOTYPE
Defects in GPR143 are the cause of ocular albinism type 1 (OA1) (Bassi, et al (1995)) (OMIM 300500) ; also known as Nettleship-Falls type ocular albinism, or X-linked Nettleship-Falls ocular albinism (XLOA). OA1 is an X-linked disorder, mainly characterized by a severe reduction in visual acuity, foveal hypoplasia, nystagmus, hypopigmentation of the retina, the presence of macromelanosomes in the skin and eyes, and the misrouting of optic pathways, resulting in the loss of stereoscopic vision.
Defects in GPR143 are a cause of late-onset sensorineural deafness (OASD) (Bassi, et al (1999)) (OMIM 300650). OASD is an X-linked recessive disorder characterized by ocular albinism and progressive sensineural hearing loss in the fourth and fifth decades of life. OASD may be caused by deletion of both GPR143/OA1 and TBL1X adjacent genes; GPR143 defects probably causing the albinism phenotype.
REFERENCE
- Bassi MT, Ramesar RS, Caciotti B, Winship IM, De Grandi A, Riboni M, Townes PL, Beighton P, Ballabio A, Borsani G. X-linked late-onset sensorineural deafness caused by a deletion involving OA1 and a novel gene containing WD-40 repeats. Am J Hum Genet 1999; 64: 1604-16. PMID: 10330347
- Bassi MT, Schiaffino MV, Renieri A, De Nigris F, Galli L, Bruttini M, Gebbia M, Bergen AA, Lewis RA, Ballabio A. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet 1995; 10: 13-9. PMID: 7647783
- d'Addio M, Pizzigoni A, Bassi MT, Baschirotto C, Valetti C, Incerti B, Clementi M, De Luca M, Ballabio A, Schiaffino MV. Defective intracellular transport and processing of OA1 is a major cause of ocular albinism type 1. Hum Mol Genet 2000; 9: 3011-8. PMID: 11115845
- Fang S, Guo X, Jia X, Xiao X, Li S, Zhang Q. Novel GPR143 mutations and clinical characteristics in six Chinese families with X-linked ocular albinism. Mol Vis. 2008; 14:1974-82. PMID: 18978956
- Iannaccone A, Gallaher KT, Buchholz J, Jennings BJ, Neitz M, Sidjanin DJ. Identification of two novel mutations in families with X-linked ocular albinism. Mol Vis 2007; 13:1856-61. PMID: 17960122
- Innamorati G, Piccirillo R, Bagnato P, Palmisano I, Schiaffino MV. The melanosomal/lysosomal protein OA1 has properties of a G protein-coupled receptor. Pigment Cell Res 2006; 19: 125-35. PMID: 16524428
- Liu JY, Ren X, Yang X, Guo T, Yao Q, Li L, Dai X, Zhang M, Wang L, Liu M, Wang QK. Identification of a novel GPR143 mutation in a large Chinese family with congenital nystagmus as the most prominent and consistent manifestation. J Hum Genet 2007; 52:565-70. PMID: 17516023
- Lopez VM, Decatur CL, Stamer WD, Lynch RM, McKay BS. L-DOPA is an endogenous ligand for OA1. PLoS Biol 2008; 6:e236. PMID: 18828673
- Micale L, Augello B, Fusco C, Turturo MG, Granatiero M, Piemontese MR, Zelante L, Cecconi A, Merla G. GPR143 mutational analysis in two Italian families with X-linked ocular albinism. Genet Test Mol Biomarkers 2009; 13: 527-31.PMID: 19604113
- Peng Y, Meng Y, Wang Z, Qin M, Li X, Dian Y, Huang S. A novel GPR143 duplication mutation in a Chinese family with X-linked congenital nystagmus. Mol Vis 2009; 15: 810-4.PMID: 19390656
- Piccirillo R, Palmisano I, Innamorati G, Bagnato P, Altimare D, Schiaffino MV. An unconventional dileucine-based motif and a novel cytosolic motif are required for the lysosomal and melanosomal targeting of OA1. J Cell Sci 2006; 119:2003-14. PMID: 16621890
- Roma C, Ferrante P, Guardiola O, Ballabio A, Zollo M. New mutations identified in the ocular albinism type 1 gene. Gene 2007; 402: 20-7. PMID: 17822861
- Schiaffino MV, Baschirotto C, Pellegrini G, Montalti S, Tacchetti C, De Luca M, Ballabio A. The ocular albinism type 1 gene product is a membrane glycoprotein localized to melanosomes. Proc Natl Acad Sci U S A 1996; 93: 9055-60. PMID: 8799153
- Schiaffino MV, d'Addio M, Alloni A, Baschirotto C, Valetti C, Cortese K, Puri C, Bassi MT, Colla C, De Luca M, Tacchetti C, Ballabio A. Ocular albinism: evidence for a defect in an intracellular signal transduction system. Nat Genet 1999; 23: 108-12. PMID: 10471510
- Shen B, Orlow SJ. The ocular albinism type 1 gene product is an N-glycoprotein but glycosylation is not required for its subcellular distribution. Pigment Cell Res 2001a; 14: 485-90. PMID: 11775061
- Shen B, Rosenberg B, Orlow SJ. Intracellular distribution and late endosomal effects of the ocular albinism type 1 gene product: consequences of disease-causing mutations and implications for melanosome biogenesis. Traffic 2001b; 2: 202-11. PMID: 11260525
- Vetrini F, Auricchio A, Du J, Angeletti B, Fisher DE, Ballabio A, Marigo V. The microphthalmia transcription factor (Mitf) controls expression of the ocular albinism type 1 gene: link between melanin synthesis and melanosome biogenesis. Mol Cell Biol 2004; 24: 6550-9. PMID: 15254223
- Wang Y, Guo XL, Wei AH, Zhu W, Li W*, Lian S*. Identification of a novel mutation in a Chinese family with X-linked ocular albinism. Eur J Ophathalmol 2009; 19: 124-8. PMID: 19123159
- Zhou P, Wang Z, Zhang J, Hu L, Kong X. Identification of a novel GPR143 deletion in a Chinese family with X-linked congenital nystagmus. Mol Vis 2008; 14: 1015-9. PMID: 18523664
EDIT HISTORY:
Created by Wei Li & Jonathan Bourne: 07/29/2004
Updated by Wei Li: 04/06/2006
Updated by Wei Li: 08/08/2007
Updated by Wei Li: 02/28/2008
Updated by Wei Li: 08/20/2008
Updated by Wei Li: 02/23/2009
Updated by Wei Li: 03/15/2010