GENOMIC
Mapping
9qD. View the map and BAC clones (data from UCSC genome browser).
Note that the Rab27a gene is located at 2.0Mb upstream.

Structure
(assembly 10/03)
Myo5a/NM_010864: 41 exons, 147,338bp, Chr9: 77,611,564-77,758,901.
The figure below shows the structure of the Myo5a gene (data from UCSC genome browser).

Regulatory Element
Search the 5'UTR and 1kb upstream regions (human and mouse) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
Myo5a (NM_010864), 7,087bp, view ORF and the alignment to genomic.
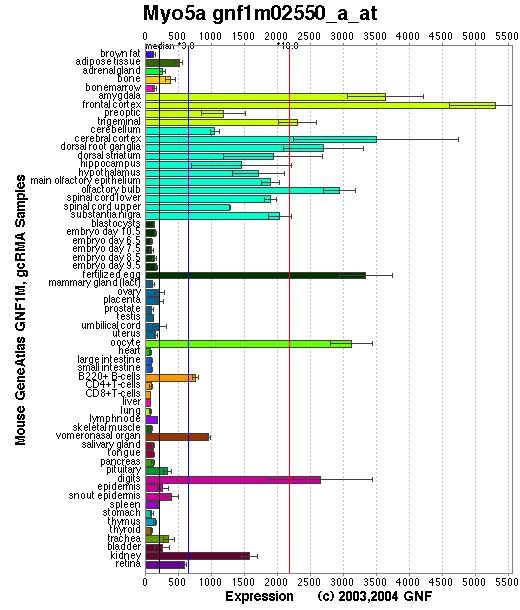
Expression Pattern
Tissue specificity: Highly expressed in melanocytes and brain. Myo5a transcripts are differentially expressed in both embryonic and adult tissues and are very abundant in neurons of the central nervous system, cephalic ganglia, and spinal ganglia (Mercer, et al).
Affymetrix microarray expression pattern in SymAtlas from GNF is shown below.
PROTEIN
Sequence
Myosin VA (NP_034994): 1853aa, ExPaSy NiceProt view of Swiss-Prot:Q99104.
Synonyms: myosin Va; dilute myosin heavy chain, non-muscle.
Ortholog
| Species | Human | Chimp | Rat | Fruitfly |
| GeneView | GS1 | 7086 | Myo5a | CG2146/didum |
| Protein | NP_000250 (1855aa) | 12113 (1885aa) | NP_071514 (1828aa) | NP_477186 (1792aa) |
| Identities | 94%/1768aa | 94%/1784aa | 97%/1800aa | 40%/751aa |
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc.
Domain
(1) Domains predicted by SMART:
a) MYSc (myosin_head): 63 - 764
b) IQ_regioin: 765 - 787, 788 - 810, 813 - 835, 836 - 858, 861 - 883, 884 - 906
c) coiled coil: 911 - 1105
d) coiled coil: 1153 - 1234
e) coiled coil: 1314 - 1364
f) coiled coil: 1406 - 1443
g) Pfam:DIL: 1685 - 1790
(2) Graphic view of the Pfam domain structure.
(3) Transmembrane domains predicted by SOSUI: None.
Motif/Site
(1) Predicted results by ScanProsite:
a) IQ motif profile :
766 - 795: score=8.773,
LRAACIRIQKTIRGWLLRKRYLCMQRAAIT
789 - 818: score=9.084,
MQRAAITVQRYVRGYQARCYAKFLRRTKAA
814 - 841: score=9.505,
RTKAATTIQKYWRMYVVRRRYKIRRAAT--
837 - 866: score=11.573,
RRAATIVIQSYLRGYLTRNRYRKILREYKA
862 - 889: score=9.304,
REYKAVIIQKRVRGWLARTHYKRTMKAI--
885 - 914: score=6.815,
TMKAIVYLQCCFRRMMAKRDVKKLKIEARS
b) Glutamic acid-rich region profile : [occurs frequently]
960 - 1265: score=8.767
c) N-glycosylation site : [occurs frequently]
156 - 159: NQSI,
216 - 219: NSSR,
651 - 654: NATT,
1120 - 1123: NESE,
1579 - 1582: NTSR,
1720 - 1723: NVSQ.
d) ATP/GTP-binding site motif A (P-loop) : [occurs frequently]
163 - 170: GesgaGKT
e) Tyrosine kinase phosphorylation site : [occurs frequently]
551 - 559: KhfaDkveY,
733 - 741: KlilDkdkY,
1197 - 1203: Rga.Ele.Y,
1380 - 1387: RltnEnl.Y.
g) Cell attachment sequence : [occurs frequently]
1545 - 1547: RGD
h) Microbodies C-terminal targeting signal : [occurs frequently]
1851 - 1853: ARV
i) Bipartite nuclear targeting sequence : [occurs frequently]
858 - 874:
RKilreykaviiqkrvr,
871 - 887:
KRvrgwlarthykrtmk.
j) Tyrosine sulfation site : [occurs frequently]
1117 - 1131:
hssneseYtfssefa,
1380 - 1394:
rltnenlYfeelyad,
1385 - 1399:
nlyfeelYaddpkky.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: none
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: too long tail
i) Dileucine motif in the tail: found: LL at 27.
3D Model
(1) ModBase matched entries found, results here.
(2)ModBase Predicted Comparative 3D Structure of Q99104 from UCSC Genome Sorter.
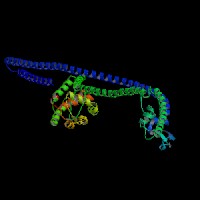

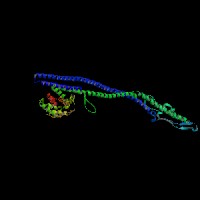
From left to right: Front, Top, and Side views of predicted protein
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=215,595Da, pI=8.84 (NP_034994).
Myosin-Va undergoes intracellular proteolysis by endogenous calpain, when synaptosomes are depolarized in the presence of calcium, making it a target for calcium signaling during synaptic activation (Casaletti, et al).
FUNCTION
Ontology
a) Biological process: cytoskeleton organization and biogenesis
b) Biological process: vesicle transport along actin filament
c) Motor activity
d) Component of myosin
e) Component of the acroplaxome of rodent spermatids
f) Actin binding; ATP binding; calmodulin binding.
Location
Cytoskeleton (actin filaments). Anti-MyoVa antibody specifically stained neuronal nuclei from forebrain to cerebellar regions, and more intensely sensory nuclei in adult Wistar rats (Telilli, et al). Immunocytochemical localization of myosin-Va in cultured hippocampal neurons shows that it partially colocalizes with PSD-95 at synapses and is also diffusely localized in cell bodies, dendrites, and axons (Walikonis, et al).
Interaction
Myosin-Va consists of two heavy chains (homodimer), each containing a neck domain with six tandem IQ motifs that bind four to five calmodulins and one to two essential light chains. The N-terminal head region consists of an actin-binding site and an ATP-binding site, with cargo-binding domain at the long neck tail region. Apo-calmodulin acts as the light chain for unconventional myosin V, and treatment with Ca(2+) can cause dissociation of calmodulin from the 6 IQ regions of the myosin heavy chain. Holo-calmodulin forms a high-affinity 1:1 complex with IQ3-IQ4 in a novel mode of interaction, as a "bridged" structure wherein two calmodulin domains interact with adjacent IQ motifs (Martin, et al).
Myosin Va binds Slac2-a/melanophilin and MyRIP ( linker proteins between Rab27A and myosin Va). Slac2-a directly interacts with Rab27a and myosin Va via its N-terminal region (amino acids 1 to 146) and the middle region (amino acids 241 to 405), respectively (Fukuda, et al). The Rab27a interacts with myosin-VIIa and myosin-Va via MyRIP or melanophilin and mediates melanosome binding to actin. The interaction between myosin Va and Rab27a is exon F dependent (Wu, et al). Myosin Va isoforms containing exon F are able to colocalize with and influence melanosome distribution by indirect interaction with rab27a and direct interaction with melanophilin. Isoforms lacking exon F but containing exon D are associated with vesicles near the Golgi area (Westbroek, et al). The MyoVa isoforms lacking exon F are not targeted to the melanosomes, but localized to the perinuclear region instead (Au, et al). The actin-activated ATPase activity of the melanocyte-type myosin Va having exon-F was significantly activated by melanophilin by 4-fold. Although Rab27a binds to myosin Va/melanophilin complex, it did not affect the melanophilin-induced activation of myosin Va. Deletion of the C-terminal actin binding domain and N-terminal Rab binding domain of melanophilin resulted in no change in the activation of the ATPase by melanophilin, indicating that the myosin Va binding domain (MBD) is sufficient for the activation of myosin Va. Among MBDs, the interaction of MBD-2 with exon-F of myosin Va is critical for the binding of myosin Va and melanophilin, whereas MBD-1 interacting with the globular tail of myosin Va plays a more significant role in the activation of myosin Va ATPase activity (Li, et al (2005)). Inhibited myosin Va is in a folded conformation such that the tail domain interacts with and inhibits myosin Va motor activity. The C-terminal globular tail domain (GTD) that directly inhibits the motor activity of myosin Va. A conserved acidic residue in the motor domain (Asp136) and two conserved basic residues in the GTD (Lys1706 and Lys1779) are critical residues for this regulation. It is proposed that binding of the GTD to the motor domain prevents the movement of the converter/lever arm during ATP hydrolysis cycle, thus inhibiting the chemical cycle of the motor domain (Li, et al (2008)).
Melanosomes move along both actin filaments (for short-range, local motility) and microtubules (for long distance movement). The leaden gene product, melanophilin, is required with Rab27a to recruit myosin Va to melanosomes in melanocytes. The reduced levels of myosin Va protein in leaden and ashen melanocytes suggests that myosin Va stability is influenced by the leaden and ashen gene products (Hume, et al). (view diagram of the Myo5a tripartite protein complex and the dynamics of melanosomes here).
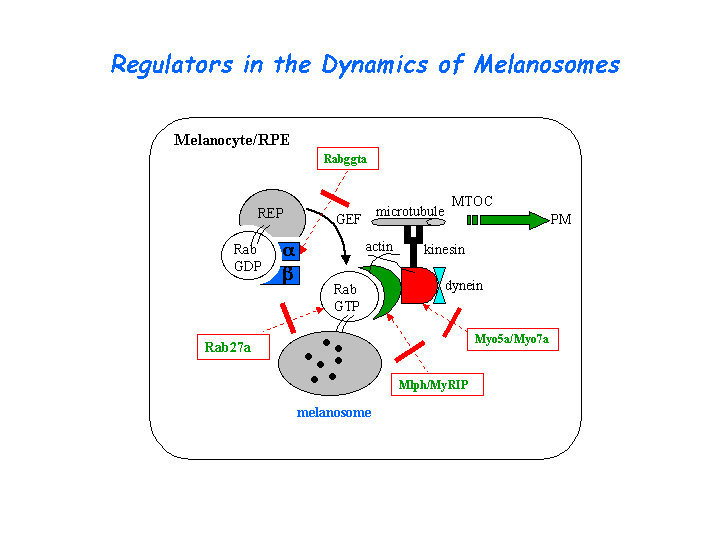{kind=link}
Rab27a and MyRIP link neuroendocrine secretory granules to F-actin and control their motion toward release sites through the actin cortex (Desnos, et al (2003)). A reduction of MyoVa protein levels achieved by RNA interference caused a significant decrease in glucose- or depolarization-stimulated insulin secretion (Varadi, et al). Myosin-Va exhibits an unusually high affinity for F-actin in the presence of ATP and induced by Ca(2+) (Tauhata, et al). The actin-based vesicle-transport motor (MyoVA) can interact directly with a microtubule-based transport motor (KhcU) through Myo5a residues 1527-1805 (Huang, et al (1999)). KHC is responsible for plus end-directed microtubule movement. On the other hand, Myo5a directly interacts with Dlc8, one of the dynein light chains (Espindola, et al). The cytoplasmic dynein acts as a minus end-directed microtubule motor. Myosin Va binds to organelles that are transported bi-directionally in axons along microtubules (Bridgman, et al). The coordination of myosin Va and microtubule-dependent motors is required for fast axonal retrograde transport in motor neurons (Lalli, et al). MyoVa inhibition reduces the number of docked granules, leading to reduced secretory responses, indicating that MyoVa directly mediates stable attachment of secretory granules at the plasma membrane (Desnos, et al (2007)). Insulin stimulation leads to Akt-mediated phosphorylation of myosin 5a at serine 1650 to enhance the interaction of myosin 5a with the actin cytoskeleton (Yoshizaki, et al).
Synaptic vesicles are one of the most important cargoes for myosin-Va. Myosin-Va on synaptic vesicles interacts with syntaxin-1A at or above 0.3 microM Ca2+. Interference with formation of the syntaxin-1A-myosin-Va complex reduces the exocytotic frequency in chromaffin cells. The syntaxin-1A-binding site was in the neck, a region that contains calmodulin-binding IQ-motifs. Syntaxin-1A binding by myosin-Va in the presence of Ca2+ depends on the release of calmodulin from the myosin-Va neck, allowing syntaxin-1A to occupy the vacant IQ-motif (Watanabe, et al). Oligodendrocytes express myosin Va and loss of myosin Va function resulted in significantly smaller lamellas and decreased process number, length, and branching of oligodendrocytes. Loss of myosin Va function also blocked distal localization of VAMP2 (Sloane, et al). Myosin Va associates with AMPARs through its cargo binding domain, which is enhanced by active GTP-bound Rab11. Myosin Va mediated the CaMKII-triggered translocation of GluR1 receptors from the dendritic shaft into spines (Correia, et al).
Myo5a drosophila homolog CG2146/didum interaction information shown in CuraGen interaction database.
Pathway
Processive actin-based motor that can move in large steps approximating the 36-nm pseudo-repeat of the actin filament. Involved in melanosome transport. May also be required for some polarization process involved in dendrite formation. Myosin-Va is a molecular motor that may participate in synaptic vesicle cycling and axonal transport.
MUTATION
Allele or SNP
39 phenotypic alleles described in MGI:105976.
SNPs deposited in dbSNP.
Distribution
The original dilute allele (dilute-viral, dv) is caused by the integration of an ecotropic murine leukemia virus (Emv-3) into the Myo5a gene located within an intron in a region of the C-terminal tail that is differentially spliced.and that phenotypic revertants of dv (termed d+) result from viral excision; a solo viral long terminal repeat (LTR) is all that remains in revertant DNA (Mercer, et al).
Huang, et al (1998a) identified the mutations responsible for 7 viable dilute alleles in the Myo5a head region. All these mutations are missense mutations, roughly equally distributed along the length of the Myo5a head. Crystallographic modeling is used to predict how these mutations might affect motor domain function. At the meantime, the same group reported 10 other mutations mapped to the coiled-coil or globular domains of the Myo5a tail region. Four of the 10 tail mutations were splicing mutations, five were missense mutations, and one resulted from an ~1kb intragenic deletion. (Huang, et al (1998b)).
The mouse neurological mutation flailer (flr) expresses a novel gene by exon shuffling that combines the promoter and first two exons of guanine nucleotide binding protein beta 5 (Gnb5) with the C-terminal exons of the closely linked Myo5a gene. The flailer protein, which is expressed predominantly in brain, contains the N-terminal 83 amino acids of Gnb5 fused in-frame with the C-terminal 711 amino acids of MyoVA, including the globular tail domain that binds organelles for intracellular transport. The flailer protein competes with wild-type MyoVA in vivo, preventing the localization of smooth endoplasmic reticulum vesicles in the dendritic spines of cerebellar Purkinje cells (Johns, et al).
Effect
The Emv-3 sequences result in the production of shortened and abnormally spliced dilute transcripts in dv mice and that the level of this effect varies among tissues. This tissue-specific effect on dilute expression likely accounts for the absence of neurological abnormalities observed in dv mice (Seperack , et al). A unique feature of the Myo5a mutations in the head region (Huang, et al (1998a)) is that they all are likely to encode proteins with residual wild-type function with multiple different phenotypic classes.
PHENOTYPE
In three pigment mutations, dilute (d), ashen (ash), and leaden (ln), melanin synthesis is normal but melanosome transport is impaired, resulting in the clumping of the melanosomes in the perinuclear region of the cell. The melanocyte phenotype of dilute, ashen, and leaden mice may be suprpressed by the dilute suppressor (dsu) gene in a semidominant manner (Moore, et al). O'Sullivan, et al (O'Sullivan, et al) identified the mutation of the dilute suppressor (dsu) gene. The wild type of dsu is predicted to encode a 214aa protein that is vertebrate specific. It does not resemble any known motor proteins or any known transcript factors that might regulate motor activity. DSU may function in a pathway separate from MYO5A. Defects in Myo5a gene are the cause of dilute (d) (Mercer, et al; Seperack , et al), an autosomal recessive disorder resembling Griscelli syndrome 1 (GS1, OMIM 214450). Hundreds of forward mutations to dilute have been identified. Most of these alleles were produced in large-scale mutagenesis screens, most of which are homozygous lethal and presumably represent null alleles. Four viable classes of alleles were also recovered and presumably represent hypomorphic alleles (Huang, et al (1998a; 1998b)). For more details of the dilute phenotype, view the Mouse Locus Catalog #Myo5a.
One class, called dilute (d), which is carried by many inbred strains of mice, produces a lightening of coat colour, causing abnormal melanocyte structure, resulting in clumping of melanin pigment in hair of nonagouti mice, producing a blue-gray coat color. This is now referred to as dilute viral (dv), which is described in more detail in JAX Mice database (Myo5adDs/J).
Most dilute-lethal alleles also produce a neurological defect, characterized by convulsions and opisthotonus, apparent at 8-10 days of age and continuing until the death of the animal at 2-3 weeks of age. Motor neurons from homozygous myosin Va null mice showed slower retrograde transport compared with wild-type cells, establishing a unique role for myosin Va in this process (Lalli, et al). Myosin Va-null mice exhibit significantly impaired myelination of the brain, optic nerve, and spinal cord. Oligodendrocytes express myosin Va and loss of myosin Va function resulted in significantly smaller lamellas and decreased process number, length, and branching of oligodendrocytes (Sloane, et al). Dilute-opisthotonus (dop) is a spontaneous gene mutation in the rat, and phenotypes of the homozygote (dop/dop) are similar to those of the Myo5a-deficient mouse. An intragenic rearrangement consisting of a 306-bp inversion associated with 17-bp and 217-bp deletions were identified in the Myo5a gene of the dop genome. This rearrangement involved a 141-bp exon, which was skipped in the dop transcript. The MyoVA protein expression was severely impaired in the dop/dop brain (Futaki , et al).
REFERENCE
- Au JS, Huang JD. A tissue-specific exon of myosin Va is responsible for selective cargo binding in melanocytes. Cell Motil Cytoskeleton 2002; 53: 89-102. PMID: 12211107
- Bridgman PC. Myosin Va movements in normal and dilute-lethal axons provide support for a dual filament motor complex. J Cell Biol 1999; 146: 1045-60. PMID: 10477758
- Casaletti L, Tauhata SB, Moreira JE, Larson RE. Myosin-Va proteolysis by Ca2+/calpain in depolarized nerve endings from rat brain. Biochem Biophys Res Commun 2003; 308: 159-64. PMID: 12890495
- Correia SS, Bassani S, Brown TC, Lis�� MF, Backos DS, El-Husseini A, Passafaro M, Esteban JA. Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat Neurosci 2008;11: 457-66. PMID: 18311135
- Desnos C, Schonn JS, Huet S, Tran VS, El-Amraoui A, Raposo G, Fanget I, Chapuis C, Menasche G, de Saint Basile G, Petit C, Cribier S, Henry JP, Darchen F. Rab27A and its effector MyRIP link secretory granules to F-actin and control their motion towards release sites. J Cell Biol 2003; 163: 559-70. PMID: 14610058
- Desnos C, Huet S, Fanget I, Chapuis C, B?ttiger C, Racine V, Sibarita JB, Henry JP, Darchen F. Myosin va mediates docking of secretory granules at the plasma membrane. J Neurosci 2007; 27: 10636-45. PMID: 17898234
- Espindola FS, Suter DM, Partata LB, Cao T, Wolenski JS, Cheney RE, King SM, Mooseker MS. The light chain composition of chicken brain myosin-Va: calmodulin, myosin-II essential light chains, and 8-kDa dynein light chain/PIN. Cell Motil Cytoskeleton 2000; 47: 269-81. PMID: 11093248
- Fukuda M, Kuroda TS, Mikoshiba K. Slac2-a/melanophilin, the missing link between Rab27 and myosin Va: implications of a tripartite protein complex for melanosome transport. J Biol Chem 2002; 277: 12432-6. PMID: 11856727
- Futaki S, Takagishi Y, Hayashi Y, Ohmori S, Kanou Y, Inouye M, Oda S, Seo H, Iwaikawa Y, Murata Y. Identification of a novel myosin-Va mutation in an ataxic mutant rat, dilute-opisthotonus. Mamm Genome 2000; 11: 649-55. PMID: 10920234
- Hume AN, Collinson LM, Hopkins CR, Strom M, Barral DC, Bossi G, Griffiths GM, Seabra MC. The leaden gene product is required with Rab27a to recruit myosin Va to melanosomes in melanocytes. Traffic 2002; 3: 193-202. PMID: 11886590
- Huang JD, Brady ST, Richards BW, Stenolen D, Resau JH, Copeland NG, Jenkins NA. Direct interaction of microtubule- and actin-based transport motors. Nature 1999; 397: 267-70. PMID: 9930703
- Huang JD, Cope MJ, Mermall V, Strobel MC, Kendrick-Jones J, Russell LB, Mooseker MS, Copeland NG, Jenkins NA. Molecular genetic dissection of mouse unconventional myosin-VA: head region mutations. Genetics 1998a; 148: 1951-61. PMID: 9560408
- Huang JD, Mermall V, Strobel MC, Russell LB, Mooseker MS, Copeland NG, Jenkins NA. Molecular genetic dissection of mouse unconventional myosin-VA: tail region mutations. Genetics 1998b; 148: 1963-72. PMID: 9560409
- Jones JM, Huang JD, Mermall V, Hamilton BA, Mooseker MS, Escayg A, Copeland NG, Jenkins NA, Meisler MH. The mouse neurological mutant flailer expresses a novel hybrid gene derived by exon shuffling between Gnb5 and Myo5a. Hum Mol Genet 2000; 9: 821-8. PMID: 10749990
- Lalli G, Gschmeissner S, Schiavo G. Myosin Va and microtubule-based motors are required for fast axonal retrograde transport of tetanus toxin in motor neurons. J Cell Sci 2003; 116: 4639-50. PMID: 14576357
- Li XD, Ikebe R, Ikebe M. Activation of myosin Va function by melanophilin, a specific docking partner of myosin Va. J Biol Chem 2005; 280: 17815-22. PMID: 15760894
- Li XD, Jung HS, Wang Q, Ikebe R, Craig R, Ikebe M. The globular tail domain puts on the brake to stop the ATPase cycle of myosin Va. Proc Natl Acad Sci U S A 2008; 105: 1140-5. PMID: 18216256
- Martin SR, Bayley PM. Regulatory implications of a novel mode of interaction of calmodulin with a double IQ-motif target sequence from murine dilute myosin V. Protein Sci 2002; 11: 2909-23. PMID: 12441389
- Mercer JA, Seperack PK, Strobel MC, Copeland NG, Jenkins NA. Novel myosin heavy chain encoded by murine dilute coat colour locus. Nature 1991; 349: 709-13. PMID: 1996138
- Moore KJ, Seperack PK, Strobel MC, Swing DA, Copeland NG, Jenkins NA. Dilute suppressor dsu acts semidominantly to suppress the coat color phenotype of a deletion mutation, dl20J, of the murine dilute locus. Proc Natl Acad Sci U S A 1988; 85: 8131-5. PMID: 3141922
- O'Sullivan TN, Wu XS, Rachel RA, Huang JD, Swing DA, Matesic LE, Hammer JA 3rd, Copeland NG, Jenkins NA. dsu functions in a MYO5A-independent pathway to suppress the coat color of dilute mice. Proc Natl Acad Sci U S A 2004; 101:16831-6. PMID: 15550542
- Seperack PK, Mercer JA, Strobel MC, Copeland NG, Jenkins NA. Retroviral sequences located within an intron of the dilute gene alter dilute expression in a tissue-specific manner. EMBO J 1995; 14: 2326-32. PMID: 7774591
- Sloane JA, Vartanian TK. Myosin Va controls oligodendrocyte morphogenesis and myelination. J Neurosci 2007; 27: 11366-75. PMID: 17942731
- Tauhata SB, dos Santos DV, Taylor EW, Mooseker MS, Larson RE. High affinity binding of brain myosin-Va to F-actin induced by calcium in the presence of ATP. J Biol Chem 2001; 276: 39812-8. PMID: 11517216
- Tilelli CQ, Martins AR, Larson RE, Garcia-Cairasco N. Immunohistochemical localization of myosin Va in the adult rat brain. Neuroscience 2003; 121: 573-86. PMID: 14568019
- Varadi A, Tsuboi T, Rutter GA. Myosin Va transports dense core secretory vesicles in pancreatic MIN6 beta-cells. Mol Biol Cell 2005; 16: 2670-80. PMID: 15788565
- Walikonis RS, Jensen ON, Mann M, Provance DW Jr, Mercer JA, Kennedy MB. Identification of proteins in the postsynaptic density fraction by mass spectrometry. J Neurosci 2000; 20: 4069-80. PMID: 10818142
- Watanabe M, Nomura K, Ohyama A, Ishikawa R, Komiya Y, Hosaka K, Yamauchi E, Taniguchi H, Sasakawa N, Kumakura K, Ushiki T, Sato O, Ikebe M, Igarashi M. Myosin-Va regulates exocytosis through the submicromolar Ca2+-dependent binding of syntaxin-1A. Mol Biol Cell 2005; 16: 4519-30. PMID: 16030255
- Westbroek W, Lambert J, Bahadoran P, Busca R, Herteleer MC, Smit N, Mommaas M, Ballotti R, Naeyaert JM. Interactions of human Myosin Va isoforms, endogenously expressed in human melanocytes, are tightly regulated by the tail domain. J Invest Dermatol 2003; 120: 465-75. PMID: 12603861
- Wu X, Wang F, Rao K, Sellers JR, Hammer JA 3rd. Rab27a is an essential component of melanosome receptor for myosin Va. Mol Biol Cell 2002; 13: 1735-49. PMID: 12006666
- Yoshizaki T, Imamura T, Babendure JL, Lu JC, Sonoda N, Olefsky JM. Myosin 5a is an insulin-stimulated Akt2 (protein kinase Bbeta) substrate modulating GLUT4 vesicle translocation. Mol Cell Biol 2007; 27: 5172-83. PMID: 17515613
EDIT HISTORY:
Created by Wei Li & Jonathan Bourne: 07/15/2004
Updated by Wei Li: 06/25/2005
Updated by Wei Li: 04/06/2006
Updated by Wei Li, 02/28/2008
Updated by Wei Li, 08/20/2008