GENOMIC
Mapping
19qC3. View the map and BAC contig (data from UCSC genome browser).
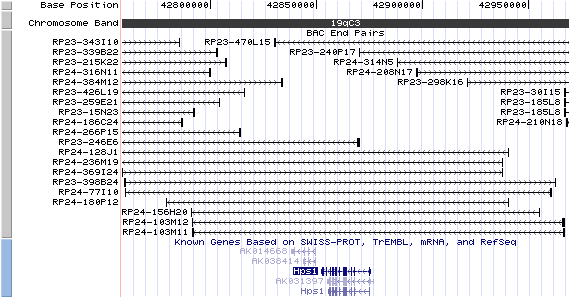
Structure
Hps1/NM_019424: 19 exons, 23,434bp, chr19:42,851,829-42,875 (assembly 10/03).
The figure below shows the structure of the Hps1 gene (data from UCSC genome browser).

Regulatory Element
Search the 5'UTR and 1kb upstream regions (seq1=human HPS1, seq2=mouse Hps1) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
Hps1/NM_019424: 2,701bp, view ORF and the alignment to genomic. Note that intron 15 contains a rare "AT-AC" nonconsensus splice site (Feng, et al (1997)) .
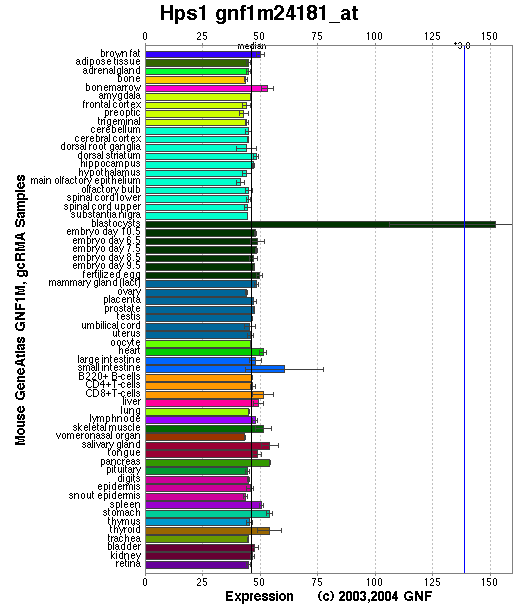
Expression Pattern
Expressed in all tissues examined with the possible exception of skeletal muscle. The highest expression was observed in lung, liver, kidney and spleen.
Affymetrix microarray expression pattern in SymAtlas from GNF is shown below.
PROTEIN
Sequence
Hps1p/Ep ( NP_062297): 704aa, ExPaSy NiceProt view of Swiss-Prot:O08983.
Synonym: Hermansky-Pudlak syndrome 1 protein homolog
Ortholog
| Species | Human | Rat | Drosophila | Mosquito | Fugu |
| GeneView | HPS1 | Hps1 | CG12855 | 1270714 | 0138156 |
| Protein | NP_000186 (700aa) | NP_414541 (706aa) | NP_610997 (596aa) | XP_309433 (602aa) | 0146520 (713aa) |
| Identities | 80%/700aa | 91%/706aa | 27%/393aa | 22%/735aa | 50%/712aa |
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc.
Domain
(1) Domains predicted by SMART:
a) coiled coil: 20-47
b) low complexity: 229-246
(2) Transmembrane domains predicted by SOSHI: none.
(3) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a) N-glycosylation site [pattern] [Warning: pattern with a high probability of occurrence]:
532 - 535 NVTM.
b) Protein kinase C phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
25 - 27 SlR, 228 - 230 TlR, 289 - 291 SsR, 351 - 353 SpR, 529 - 531 SrR.
c) N-myristoylation site [pattern] [Warning: pattern with a high probability of occurrence]:
325 - 330 GTppSD, 416 - 421 GqeaGS, 643 - 648 GVlgGD.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: none
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: none
i) Dileucine motif in the tail: none
3D Model
(1) ModBase: none.
(2) 3D models predicted by SPARKS (fold recognition) below. View the models by PDB2MGIF.
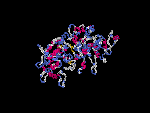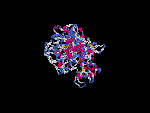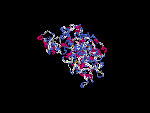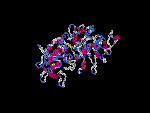
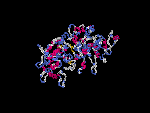
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=79,853Da, pI=5.49 (O08983).
(1) Domains predicted by SMART:
a) coiled coil: 20-47
b) low complexity: 229-246
(2) Transmembrane domains predicted by SOSHI: none.
(3) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite:
a) N-glycosylation site [pattern] [Warning: pattern with a high probability of occurrence]:
532 - 535 NVTM.
b) Protein kinase C phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
25 - 27 SlR, 228 - 230 TlR, 289 - 291 SsR, 351 - 353 SpR, 529 - 531 SrR.
c) N-myristoylation site [pattern] [Warning: pattern with a high probability of occurrence]:
325 - 330 GTppSD, 416 - 421 GqeaGS, 643 - 648 GVlgGD.
(2) Predicted results of subprograms by PSORT II:
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: none
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: none
i) Dileucine motif in the tail: none
3D Model
(1) ModBase: none.
(2) 3D models predicted by SPARKS (fold recognition) below. View the models by PDB2MGIF.
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=79,853Da, pI=5.49 (O08983).
FUNCTION
Ontology
a) Biological process: melanocyte differentiation
b) Biological process: organelle organization and biogenesis
c) Component of multiple cytoplasmic organelles.
d) Apparently crucial for organellar development and function.
e) May be involved in intracellular protein sorting.
Location
Cytoplasm, may be associated with membrane fraction.
Interaction
Hps1/ep is a component of a protein complex termed biogenesis of lysosome-related organelles complex 3 (BLOC-3), where Hps4/le is residing as another subunit (Chiang, et al; Martina, et al; Nazarian, et al) (view diagram of BLOC-3 complex here).
{kind=link}
No Hps1 drosophila homolog CG12855 interaction information shown in CuraGen interaction database.
Pathway
Hps1/ep may play a role in the early stages of the maturation of melanosomes (Nguyen, et al (2002)) (view diagram of melanosome blockage here). The mechanism is distinct from that dependent on the AP-3 complex (Dell'Angelica, et al; Feng, et al (2002)) (view diagram of BLOC-3 and AP-3 pathway here).
{kind=link}
{kind=link}
MUTATION
Allele or SNP
2 phenotypic alleles described in MGI:2177763.
SNPs deposited in dbSNP.
Distribution
| Location | Genomic | cDNA | Protein | Type | Strain | Reference |
| Exon 17 | 1835-1858del 23bp, insTGT | 1835-1858del 23bp, insTGT | R612-F619 del 23bp, insTGT | frame-shift, 613X | ep6J (B6) | Feng, et al (1997) |
| Exon 19 | 1974ins ~5.3kb IAP, 1969-1974dup | 1974ins 630bp IAP, 1969-1974dup | S658ins 630bp IAP | in-frame,78aa replaces 46aa of wild-type at C-terminus | ep (B6) | Gardner, et al |
(Numbering of genomic and cDNA sequence is based on the start codon of RefSeq NM_019424.)
Effect
The mutant transcripts of the Hps1/ep gene were observed in the Northern blots (Gardner, et al ). The ep6J allele is predicted to be a null mutation. Both ep alleles alter the coding frame and result in truncation or elongation of the Hps1 proteins.
PHENOTYPE
The recessive mutation at the pale ear (ep) locus is the homologue of human HPS1 (Feng, et al (1997); Gardner et al). The mice with the ep mutation exhibit abnormalities similar to human HPS (OMIM 203300) patients in melanosomes and platelet-dense granules (Novak, et al). The strain is described in more detail in JAX Mice database (B6.C3Fe-Hps1ep/J). Homozygotes for spontaneous mutations exhibit hypopigmentation, a platelet defect, impaired natural killer cell function, lung abnormalities, reduced secretion of kidney lysosomal enzymes, and abnormal retinofugal neuronal projections (for more details of the ep phenotype, please see the Mouse Locus Catalog #Hps1). The gene mutation in ep mice reveals a differential regulation of melanocyte function in dorsal back skin melanocytes versus tail or ear skin. In the tail, the defective gene causes delayed onset of interfollicular epidermal melanocyte tyrosinase activity, decreased numbers of melanocytes in the interfollicular epidermis and dermis, and severe immaturity of tail epidermal melanosomes, which are not observed in dorsal back follicular melanocytes. This indicates that Hps1/ep plays a developmental role in determining interfollicular epidermal and dermal melanocyte function (Nguyen, et al (2007) ).
Giant lamellar body degeneration (GLBD) was found in both ep and bg mice soon after birth, and increased in severity as the mice grew older. Aged ep mice with less severe GLBD than bg mice of comparable ages also had a slight tendency to develop interstitial inflammation but no fibrosis. Ep and bg mice, especially the latter, may be useful mouse models of HPS-associated interstitial pneumonia (HPSIP) (Tang, et al).The ep mouse has a phenotype identical to light ear (le or Hps4) (Suzuki, et al). Very few melanosomes of the retinal pigment epithelium (RPE) are observed in the BLOC-3 mutants Hps1/ep and Hps4/le. A unique feature of ep and le is the presence of very large macromelanosomes within the choroids (Gardner, et al; Suzuki, et al). In addition, ep/ep-le/le doubly homozygous mouse presents a phenotype identical to that of the singly homozygous mutants (Meisler, et al). Mice doubly homozygous for the pale ear and ruby eye had the shortest life spans with none surviving the two-year experimental duration (McGarry, et al).
Lyerla et al demonstrated a mouse model of HPS, which is homozygously recessive for both the Hps1 (pale ear) and Hps2 (pearl) genes (Feng, et al (2002)), exhibits striking abnormalities of lung type II cells. Type II cells and lamellar bodies of this mutant are greatly enlarged, and the lamellar bodies are engorged with surfactant. Giant lamellar bodies (GLB) formation is not associated with abnormal trafficking or recycling of surfactant material. Instead, impaired secretion is an important component of GLB formation in ep/pe mice (Guttentag, et al). HPS double mutant ep/pe mouse strain develops interstitial pneumonia (HPSIP) past 1 year of age, which may be initiated by abnormal ATII cells and exacerbated by alveolar macrophage activation with elevated level of TGFbeta1 (Wang and Lyerla). Inflammation is initiated from the abnormal alveolar epithelial cells in ep/pe double mutant, and S-nitrosylated SP-D plays a significant role in amplifying pulmonary inflammation (Atochina-Vasserman, et al). Aberrant surfactant trafficking and secretion may lead to the apoptosis of alveolar epithelial type II cell in HPSIP, thereby causing the development of HPSIP (Mahavadi,et al). Mutant lungs accumulate excessive autofluorescent pigment. The air spaces of mutant lungs contain age-related elevations of inflammatory cells and foamy macrophages. In vivo measurement of lung hysteresivity demonstrated aberrant lung function in mutant mice. All these features are similar to the lung pathology described in HPS patients. Morphometry of mutant lungs indicates a significant emphysema. These mutant mice provide a model to further investigate the lung pathology and therapy of HPS (Lyerla et al).
REFERENCE
- Atochina-Vasserman EN, Bates SR, Zhang P, Abramova H, Zhang Z, Gonzales L, Tao JQ, Gochuico BR, Gahl W, Guo CJ, Gow AJ, Beers MF, Guttentag S. Early alveolar epithelial dysfunction promotes lung inflammation in a mouse model of Hermansky-Pudlak syndrome. Am J Respir Crit Care Med 2011; 184: 449-58.PMID: 21616998
- Chiang PW, Oiso N, Gautam R, Suzuki T, Swank RT, Spritz RA. The Hermansky-Pudlak syndrome 1 (HPS1) and HPS4 proteins are components of two complexes, BLOC-3 and BLOC-4, involved in the biogenesis of lysosome-related organelles. J Biol Chem 2003; 278: 20332-7. PMID: 12663659
- Dell'Angelica EC, Aguilar RC, Wolins N, Hazelwood S, Gahl WA, Bonifacino JS. Molecular characterization of the protein encoded by the Hermansky-Pudlak syndrome type 1 gene. J Biol Chem 2000; 275: 1300-6. PMID: 10625677
- Feng GH, Bailin T, Oh J, Spritz RA. Mouse pale ear (ep) is homologous to human Hermansky-Pudlak syndrome and contains a rare 'AT-AC' intron. Hum Mol Genet 1997; 6: 793-7. PMID: 9158155
- Feng L, Novak EK, Hartnell LM, Bonifacino JS, Collinson LM, Swank RT. The Hermansky-Pudlak syndrome 1 (HPS1) and HPS2 genes independently contribute to the production and function of platelet dense granules, melanosomes, and lysosomes. Blood 2002; 99: 1651-8. PMID: 11861280
- Gardner JM, Wildenberg SC, Keiper NM, Novak EK, Rusiniak ME, Swank RT, Puri N, Finger JN, Hagiwara N, Lehman AL, Gales TL, Bayer ME, King RA, Brilliant MH. The mouse pale ear (ep) mutation is the homologue of human Hermansky-Pudlak syndrome. Proc Natl Acad Sci U S A 1997; 94: 9238-43. PMID: 9256466
- Guttentag SH, Akhtar A, Tao JQ, Atochina E, Rusiniak ME, Swank RT, Bates SR. Defective surfactant secretion in a mouse model of hermansky-pudlak syndrome. Am J Respir Cell Mol Biol 2005; 33:14-21. PMID: 1579097
- Lyerla TA, Rusiniak ME, Borchers M, Jahreis G, Tan J, Ohtake P, Novak EK, Swank RT. Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am J Physiol Lung Cell Mol Physiol 2003; 285: L643-53. PMID: 12777251
- Mahavadi P, Korfei M, Henneke I, Liebisch G, Schmitz G, Gochuico BR, Markart P, Bellusci S, Seeger W, Ruppert C, Guenther A. Epithelial stress and apoptosis underlie Hermansky-Pudlak syndrome-associated interstitial pneumonia. Am J Respir Crit Care Med 2010;182: 207-19. PMID: 20378731
- Martina JA, Moriyama K, Bonifacino JS. BLOC-3, a protein complex containing the Hermansky-Pudlak syndrome gene products HPS1 and HPS4. J Biol Chem 2003; 278: 29376-84. PMID: 12756248
- McGarry MP, Reddington M, Novak EK, Swank RT. Survival and lung pathology of mouse models of Hermansky-Pudlak syndrome and Chediak-Higashi syndrome. Proc Soc Exp Biol Med. 1999; 220: 162-8. PMID: 10193444
- Meisler MH, Wanner L, Strahler J. Pigmentation and lysosomal phenotypes in mice doubly homozygous for both light-ear and pale-ear mutant alleles. J Hered 1984; 75: 103-6. PMID: 6232310
- Nazarian R, Falcon-Perez JM, Dell'Angelica EC. Biogenesis of lysosome-related organelles complex 3 (BLOC-3): a complex containing the Hermansky-Pudlak syndrome (HPS) proteins HPS1 and HPS4. Proc Natl Acad Sci USA 2003; 100: 8770-5. PMID: 12847290
- Nguyen T, Novak EK, Kermani M, Fluhr J, Peters LL, Swank RT, Wei ML. Melanosome morphologies in murine models of hermansky-pudlak syndrome reflect blocks in organelle development. J Invest Dermatol 2002; 119: 1156-64. PMID: 12445206
- Nguyen T, Wei ML. Hermansky-Pudlak HPS1/pale ear gene regulates epidermal and dermal melanocyte development.J Invest Dermatol 2007 ; 127:421-8.PMID: 17068483
- Novak EK, Hui SW, Swank RT. The mouse pale ear pigment mutant as a possible animal model for human platelet storage pool deficiency. Blood 1981; 57: 38-43. PMID: 7448413
- Suzuki T, Li W, Zhang Q, Karim A, Novak EK, Sviderskaya EV, Hill SP, Bennett DC, Levin AV, Nieuwenhuis HK, Fong CT, Castellan C, Miterski B, Swank RT, Spritz RA. Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet 2002; 30: 321-4. PMID: 11836498
- Tang X, Yamanaka S, Miyagi Y, Nagashima Y, Nakatani Y. Lung pathology of pale ear mouse (model of Hermansky-Pudlak syndrome 1) and beige mouse (model of Chediak-Higashi syndrome): severity of giant lamellar body degeneration of type II pneumocytes correlates with interstitial inflammation. Pathol Int 2005; 55:137-43.PMID: 15743322
- Wang L, Lyerla T. Histochemical and cellular changes accompanying the appearance of lung fibrosis in an experimental mouse model for Hermansky Pudlak syndrome. Histochem Cell Biol 2010; 134: 205-13. PMID: 20603711
EDIT HISTORY:
Created by Wei Li: 05/26/2004
Updated by Wei Li: 06/25/2005
Updated by Wei Li: 10/08/2007
Updated by Wei Li: 06/28/2012