GENOMIC
Mapping
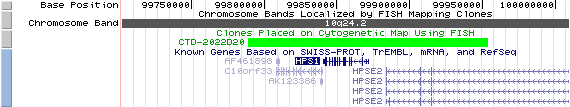
10q24.2. View the map and BAC clones of FISH (data from UCSC genome browser).
Structure
(assembly 07/03)
Isoform a (NM_000195): 20 exons, 30,750bp, chr10:99,840,542-99,871,291.
Isoform b (NM_182637): 19 exons, 30,750 bp, chr10:99,840,542-99,871,291.
Isoform c (NM_182939): 10 exons, 17,801 bp, chr10:99,853,491-99,871,291.
Isoform d (NM_182638): 17 exons, 30,750 bp, chr10:99,840,542-99,871,291.
1) NM_182637 uses an alternative splicing donor of exon 6 by extending 43bp to intron 6, which results in a frameshift and a stop codon on 195. The resulting 194aa protein (isoform b) has a distinct C-terminus and is shorter than isoform (a) which is predicted as non-functional due to permanently suppression as a nonsense-mediated mRNA decay (NMD) candidate.
2) NM_182639 skips the splicing donor of intron 10 and uses an alternate 3' coding region compared to NM_000195. It encodes isoform (c), which has a shorter and distinct C-terminus compared to isoform (a).
3) NM_182638 skips exons 5, 7 and 9 in the coding region compared to NM_000195. It encodes the shorter isoform (d), which is permanently suppressed because it is a nonsense-mediated mRNA decay (NMD) candidate.
The figure shows the comparison of these four isoforms (data from UCSC genome browser).

Regulatory Element
Search the 5'UTR and 1kb upstream regions (seq1=human HPS1, seq2=mouse Hps1) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
a) Transcript variant 1 (NM_000195), 3,714bp, view ORF and the alignment to genomic. Note that intron 16 contains a rare "AT-AC" nonconsensus splice site.
b) Transcript variant 2 (NM_182637), 3,667bp, view ORF and the alignment to genomic. Note that intron 15 contains a rare "AT-AC" nonconsensus splice site.
c) Transcript variant 3 (NM_182639), 1,625bp, view ORF and the alignment to genomic.
d) Transcript variant 4 (NM_182638), 3,320bp, view ORF and the alignment to genomic. Note that intron 13 contains a rare "AT-AC" nonconsensus splice site.
Expression Pattern
Tissue specificity: ubiquitous. On Northern blot analysis, the standard transcript is 3.0kb. Minor 3.9kb and 4.4kb mRNAs are apparent (Oh, et al (1996)). An additional 1.5kb transcript (variant 3) was found in bone marrow and melanoma cells (Wildenberg et al).
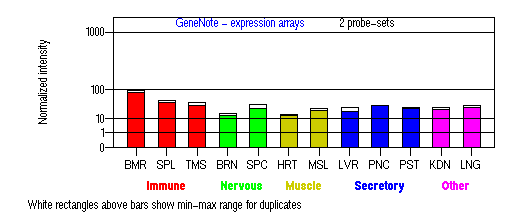
BMR: Bone marrow; SPL: Spleen; TMS: Thymus; BRN: Brain; SPC: Spinal cord; HRT: Heart; MSL: Skeletal muscle; LVR; Liver; PNC: Pancreas; PST: Prostate; KDN: Kidney; LNG: Lung. (data from GeneCards )
View more information in Entrez Gene, or UCSC Gene Sorter, or GeneCards.
PROTEIN
Sequence
(HPS1p)
Isoform a ( NP_000186): 700aa, ExPaSy NiceProt view of Swiss-Prot:Q92902.
Isoform b ( NP_872575): 194aa.
Isoform c ( NP_872577): 324aa.
Isoform d ( NP_872576): 195aa.
Synonym: Hermansky-Pudlak syndrome 1 protein.
Ortholog
(Isoform a)
| Species | Mouse | Rat | Fugu | Drosophila | Mosquito |
| GeneView | ep/Hps1 | Hps1 | 0138156 | CG12855 | 1270714 |
| Protein | NP_062297 (704aa) | NP_414541 (706aa) | 0146520 (713aa) | NP_610997 (596aa) | XP_309433 (602aa) |
| Identities | 76%/704aa | 78%/706aa | 51%/717aa | 37%/187aa | 25%/371aa |
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc.
Domain
(1) Domains predicted by SMART(isoform a):
a) coiled coil: 31-47
b) low complexity: 229-244
c) low complexity: 275-286
(2) Transmembrane domains predicted by SOSHI(isoform a): none.
(3) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite (isoform a):
a) N-glycosylation site [pattern] [Warning: pattern with a high probability of occurrence]:
528 - 531 NITM, 560 - 563 NCSQ.
b) Protein kinase C phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
25 - 27 SlR, 228 - 230 SlR, 258 - 260 SpR, 418 - 420, SlR, 525 - 527 SrR, 562 - 564 SqK.
c) N-myristoylation site [pattern] [Warning: pattern with a high probability of occurrence]:
31 - 36 GqseNE, 202 - 207 GgeeAL, 283 - 288 GgssAE, 639 - 644 GMlgGD.
(2) Predicted results of subprograms by PSORT II(isoform a):
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: none
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: none
i) Dileucine motif in the tail: none
3D Model
(1) ModBase: none.
(2) 3D models of isoform (a) predicted by SPARKS (fold recognition) below. View the models by PDB2MGIF.
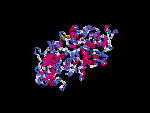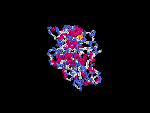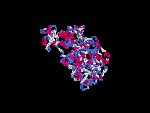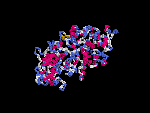
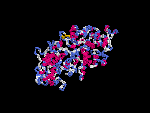
2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=79,320Da, pI=5.68 (Q92902, isoform a).
Computed theoretical MW=22,153Da, pI=4.66 (isoform b).
Computed theoretical MW=36,476Da, pI=4.74 (isoform c).
Computed theoretical MW=21,196Da, pI=6.14 (isoform d).
FUNCTION
Ontology
a) Biological process: lysosome organization and biogenesis.
b) Component of multiple cytoplasmic organelles.
c) Maturation or structure of cytoplasmic organelles, i.e. melanosomes, platelet dense bodies, lysosomes. Likely involved in assembly of these organelles.
d) Involved in the biogenesis of early melanosomes and the sorting of tyrosinase and Tyrp1 (view diagram of melanosome maturation and melanosomal protein sorting here).
{kind=link}
Location
Cytoplasm, may be associated with membrane fraction.
The HPS1 protein (HPS1p) was ariginally predicted to be an integral membrane protein having two transmembrane domains (residues 79-95 and 369-396). However, subsequent biochemical characterization using specific antibodies revealed that endogenous HPS1p from human and mouse cells exists as both cytosolic and peripheral membrane proteins. Immunoelectron microscopy analyses of HPS1p in melanoma-derived cell lines or non-melanogenic cell lines have shown that the protein is associated with the trans-Golgi network (Starcevic, et al). Membranous complexes of HPS-1 melanocytes are macroautophagosomal representatives of the lysosomal compartment (Smith, et al).
Interaction
HPS1 is a component of a protein complex termed biogenesis of lysosome-related organelles complex 3 (BLOC-3), where HPS4 is residing as another subunit (Chiang, et al; Martina, et al; Nazarian, et al). The BLOC-3 complex is a moderately asymmetric complex with a molecular mass of about 175 kD (view diagram of BLOC-3 complex here). The BLOC-3 complex dissociates into smaller complex upon Tris treatment and a portion of HPS1 exists in a cytosolic complex that does not contain HPS4 (Chiang, et al). Two regions in HPS1, spanning amino acids 1-249 and 506-700 are required for binding to HPS4 (residues 340-528)(Carmona-Rivera, et al (2013)). Interaction of BLOC-3 with the GTP-bound form of Rab9 is mediated by HPS4 and the switch I and II regions of Rab9, suggesting that BLOC-3 might function as a Rab9 effector in the biogenesis of LROs (Kloer, et al). BLOC-3 is a Rab32 and Rab38 guanine nucleotide exchange factor (GEF), to promote specific membrane recruitment of Rab32/38 (Gerondopoulos, et al).
{kind=link}
View interactions in HPRD
View co-occured partners in literature searched by PPI Finder.
Pathway
HPS1 may play a role in organelle biogenesis associated with melanosomes, platelet dense granules, and lysosomes. The mechanism is distinct from that dependent on the AP-3 complex (Dell'Angelica, et al, Feng, et al). HPS4 and HPS1 proteins may function in the same pathway of organelle biogenesis (Suzuki, et al (2002)) (view diagram of BLOC-3 pathway here). In mutant cells lacking BLOC-3, the percentages of cells displaying pronounced perinuclear accumulation were reduced (Nazarian, et al; Falcon-Perez, et al). A relatively lower frequency of microtubule-dependent movement events, either toward or away from the perinuclear region were observed in BLOC-3 deficient cells. This suggests that BLOC-3 is required for optimal attachment of late endosomes to microtubule-dependent motors (Falcon-Perez, et al). BLOC-3 defines a novel Rab GEF family with a specific function in the biogenesis of lysosome-related organelles (Gerondopoulos, et al).
{kind=link}
MUTATION
Allele or SNP
23 mutations deposited in HGMD.
SNPs deposited in dbSNP.
7 selected allelic examples described in OMIM.
Distribution
30 alleles to date.
| Location | Genomic | cDNA | Protein | Type | Ethnicity | Reference |
| Exon 3 | 94_97 delCAGT | 94_97 delCAGT | Q32delCAGT | frame-shift 50X | Germany | Sandrock, et al |
| Exon 4 | 163_165 delATC | 163_165 delATC | I55delATC | in-frame | Afghan | Oh, et al (1998) |
| Exon 5 | 288delG | 288delG | G96delT | frame-shift 123X | Japanese | Spritz, et al |
| Exon 5 | 355delC | 355delC | H119delC | frame-shift 123X | German Polish | Hermos, et al |
| Exon 5 | 391C>T | 391C>T | R131X | nonsense | Spanish Caucasian Chinese | Hermos, et al; Wei, et al (2011) |
| Exon 5 | 397G>T | 397G>T | E133X | nonsense | Italian German Ukrainian | Shotelersuk, et al |
| Intron 5 | splicing donor +5G>A | 398+5G>A 256_398del (exon 5) | F86del 143bp | splicing 133X | Japanese Indian | Oh, et al (1998); Suzuki, et al (2004); Vincent, et al |
| Exon 6 | 418delG | 418delG | A140delG | frame-shift 184X | Spanish Irish English | Hermos, et al |
| Exon 6 | 467_476del | 467_476del | Y156del 10bp | frame-shift 161X | Honduran- Salvadoran | Carmona-Rivera, et al (2011a) |
| Exon 6 | 507G>A | 507G>A | E169ins 43bp | splicing 195X | African American | Merideth, et al |
| Intron 6 | splicing donor +1G>A | 507+1G>A | E169ins 43bp | splicing 195X | Japanese | Natsuga, et al |
| Exon 7 | 532insC | 532insC | Q178insC | frame-shift 181X | Japanese | Ito, et al; Iwakawa, et al |
| Exon 8 | 716T>C | 716T>C | L239P | missense | Irish German | Hermos, et al |
| Exon 10 | 937G>A | 937G>A | G313S | missense cryptic splicing frame-shift,323X | Perto Rican | Carmona-Rivera, et al (2011b) |
| Exon 11 | 962delG | 962delG | G321delG | frame-shift 330X | Ukrainian | Oh, et al (1998) |
| Exon 11 | 962insG | 962insG | G321insG | frame-shift 452X | Japanese | Hermos, et al |
| Exon 11 | 965delC | 965delC | T322delC | frame-shift 330X | Japanese African-American | Oh, et al (1998) Merideth, et al |
| Exon 11 | 965insC | 965insC | T322insC | frame-shift 452X | Swiss Irish German French Scottish Chinese | Oh, et al (1998) Wei, et al (2010) Wei, et al (2011) |
| Exon 11 | 972delC | 972delC | M325delC | frame-shift 330X | Mexican | Carmona-Rivera, et al (2011a) |
| Intron 11 | splicing acceptor -1G>T | c.988-1G>T del 168bp (exon 12) | I330del 168bp | splicing del 56aa | Indian | Vincent, et al |
| Exon 13 | 1189delC | 1189delC | E397delC | frame-shift 398X | Caucasian | Oh, et al (1998) |
| Exon 13 | 1325insA | 1325insA | A441insA | frame-shift 452X | Japanese | Oh, et al (1996) |
| Exon 14 | 1375delA 1388C>A | 1375delA 1388C>A | S459delA S463Y | frame-shift 474X missense | English Scottish Irish | Hermos, et al |
| Exon 15 | 1471_1487dup | 1471_1487dup | P496ins 16bp | frame-shift 587X | Puerto Rican | Oh, et al (1996) |
| Exon 17 | 1691delA | 1691delA | K564delA | frame-shift 585X | Japanese | Ito, et al |
| Intron 17 | splicing acceptor -2A>C | ? | ? | splicing | Caucasian | Oetting, et al |
| Exon 18 | 1749G>A | 1749G>A | W583X | nonsense | Japanese | Ito, et al |
| Exon 19 | 1885delC | 1885delC | P629delC | frame-shift 724X | Chinese | Wei, et al (2011) |
| Exon 19 | 1932delC | 1932delC | D644delC | frame-shift 724X | Chinese | Wei, et al (2009) |
| Exon 20 | 1996G>T | 1996G>T | E666X | nonsense | Scottish | Oh, et al (1998) |
| Exon 20 | 2003T>C | 2003T>C | L668P | missense | Japanese | Ito, et al |
(Numbering of genomic and cDNA sequence is based on the start codon of RefSeq NM_000195. View ORF here.)
The most common mutation is the 16bp duplication within exon 15 (founder mutation) in northwest Puerto Rican patients. In Japanese, the IVS5 +5G>A mutation appears to represent a founder effect (Ito, et al). A frameshift hot spot at codons 321-322 is apparent in non-Puerto Rican patients (Oh, et al (1998)). Exons 5, 11, and 15 are apparently hotspots for mutation screening.
Note that a pseudogene has been detected on chromosome 22q12.2-12.3 which has high sequence similarity to HPS1 exons 2-5 and 100% sequence homology to HPS1 exon 6. Careful check should be alerted when base pair change is observed in these regions.
Effect
Most of the HPS1 gene mutations are frameshift mutations or nonsense mutations that disrupt the function of the HPS1 protein. Five splicing mutations have been reported including the c.507G>A mutation. Two missense mutations (L239P and L668P) have been reported. L239 residue is conserved in different species (refer to the multiple sequence alignment PDF file). Overexpression of L668P mutant HPS1 protein in le-melanocytes did not restore the stability of endogeneous HPS4, a subunit of BLOC-3 (Ito, et al).
Interestingly, both the silence mutation (c.507G>A) and the splicing mutation (c.507+1G>A) lead to the activation of a cryptic splice site at +43 bp within intron 6 of the HPS1 gene, which results in a frameshift of the mutant transcript, predicting that it will be targeted for nonsense-mediated decay or that it will produce a truncated protein with a premature stop codon 25 amino acids downstream of E169E (Merideth, et al; Netsuga, et al), which is the same as isoform (b).
The c.1932delC mutation leads to a longer HPS1 protein that a novel 79-residue peptide replaces the wild-type 56-residue peptide after the mutation site at D644 (Wei, et al (2009)). However, the elongated HPS1 protein is unstable in the patient's platelets (Wei and Li.).
PHENOTYPE
Hermansky-Pudlak syndrome (HPS, OMIM 203300) was first described by Hermansky and Pudlak (1959). It is a rare autosomal recessive disorder in which oculocutaneous albinism, bleeding tendency, and lysosomal ceroid storage result from defects of multiple cytoplasmic organelles: melanosomes, platelet-dense granules, and lysosomes. Mutations in this gene are associated with Hermansky-Pudlak syndrome type 1 (HPS-1, OMIM 604982). HPS-1 is the most common HPS type.
Some HPS-1 adult patients developed pulmonary fibrosis and granulomatous colitis (Hermos et al). These complications are common among Puerto Rican HPS-1 patients but have not appeared in HPS-2 or HPS-3 patients. Oh et al (1996) found that the different clinical HPS phenotypes were associated with different HPS1 frameshifts, which suggested that differentially truncated HPS1 polypeptides may have somewhat different consequences for subcellular function. In some HPS1 patients, no apparent bleeding tendency were noticed, suggesting the severity of HPS1 is associated with the genotype (Wei, et al (2009, 2010)).
REFERENCE
- Carmona-Rivera C, Golas G, Hess RA, Cardillo ND, Martin EH, O'Brien K, Tsilou E, Gochuico BR, White JG, Huizing M, Gahl WA. Clinical, molecular, and cellular features of non-Puerto Rican Hermansky-Pudlak syndrome patients of Hispanic descent. J Invest Dermatol 2011a; 131: 2394-400. PMID: 21833017
- Carmona-Rivera C, Hess R, O'Brien K, Golas G, Tsilou E, White J, Gahl W, Huizing M. Novel mutations in the HPS1 gene among Puerto Rican patients. Clin Genet 2011b; 79: 561-7. PMID: 20662851
- Carmona-Rivera C, Simeonov DR, Cardillo ND, Gahl WA, Cadilla CL. A divalent interaction between HPS1 and HPS4 is required for the formation of the biogenesis of lysosome-related organelle complex-3 (BLOC-3). Biochim Biophys Acta 2013; 1833: 468-78. PMID: 23103514
- Chiang PW, Oiso N, Gautam R, Suzuki T, Swank RT, Spritz RA. The Hermansky-Pudlak syndrome 1 (HPS1) and HPS4 proteins are components of two complexes, BLOC-3 and BLOC-4, involved in the biogenesis of lysosome-related organelles. J Biol Chem 2003; 278: 20332-7. PMID: 12663659
- Dell'Angelica EC, Aguilar RC, Wolins N, Hazelwood S, Gahl WA, Bonifacino JS. Molecular characterization of the protein encoded by the Hermansky-Pudlak syndrome type 1 gene. J Biol Chem 2000; 275: 1300-6. PMID: 10625677
- Falcon-Perez JM, Nazarian R, Sabatti C, Dell'angelica EC. Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J Cell Sci 2005; 118: 5243-55.PMID: 16249233
- Feng L, Novak EK, Hartnell LM, Bonifacino JS, Collinson LM, Swank RT. The Hermansky-Pudlak syndrome 1 (HPS1) and HPS2 genes independently contribute to the production and function of platelet dense granules, melanosomes, and lysosomes. Blood 2002; 99: 1651-8. PMID: 11861280
- Gerondopoulos A, Langemeyer L, Liang JR, Linford A, Barr FA. BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr Biol 2012; 22: 2135-9. PMID: 23084991
- Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood 1959; 14: 162-9. (No Abstract)
- Hermos CR, Huizing M, Kaiser-Kupfer MI, Gahl WA. Hermansky-Pudlak syndrome type 1: gene organization, novel mutations, and clinical-molecular review of non-Puerto Rican cases. Hum Mutat 2002; 20:482. PMID: 12442288
- Ito S, Suzuki T, Inagaki K, Suzuki N, Takamori K, Yamada T, Nakazawa M, Hatano M, Takiwaki H, Kakuta Y, Spritz RA, Tomita Y.High frequency of Hermansky-Pudlak syndrome type 1 (HPS1) among Japanese albinism patients and functional analysis of HPS1 mutant protein. J Invest Dermatol 2005; 125:715-20.PMID: 16185271
- Iwakawa J, Matsuyama W, Watanabe M, Yamamoto M, Oonakahara K, Machida K, Higashimoto I, Niiyama T, Osame M, Arimura K. Hermansky-Pudlak syndrome with a novel mutation. Intern Med 2005; 44:733-8. PMID: 16093596
- Kloer DP, Rojas R, Ivan V, Moriyama K, van Vlijmen T, Murthy N, Ghirlando R, van der Sluijs P, Hurley JH, Bonifacino JS. Assembly of the biogenesis of lysosome-related organelles complex-3 (BLOC-3) and its interaction with Rab9. J Biol Chem 2010; 285: 7794-804.PMID: 20048159
- Martina JA, Moriyama K, Bonifacino JS. BLOC-3, a protein complex containing the Hermansky-Pudlak syndrome gene products HPS1 and HPS4. J Biol Chem 2003; 278: 29376-84. PMID: 12756248
- Merideth MA, Vincent LM, Sparks SE, Hess RA, Manoli I, O'Brien KJ, Tsilou E, White JG, Huizing M, Gahl WA. Hermansky-Pudlak syndrome in two African-American brothers. Am J Med Genet A 2009; 149A:987-92. PMID: 19334085
- Natsuga K, Akiyama M, Shimizu T, Suzuki T, Ito S, Tomita Y, Tanaka J, Shimizu H. Ultrastructural features of trafficking defects are pronounced in melanocytic nevus in Hermansky-Pudlak syndrome type 1. J Invest Dermatol 2005; 125:154-8. PMID: 15982315
- Nazarian R, Falcon-Perez JM, Dell'Angelica EC. Biogenesis of lysosome-related organelles complex 3 (BLOC-3): a complex containing the Hermansky-Pudlak syndrome (HPS) proteins HPS1 and HPS4. Proc Natl Acad Sci USA 2003; 100: 8770-5. PMID: 12847290
- Oetting WS, King RA. Molecular basis of albinism: mutations and polymorphisms of pigmentation genes associated with albinism. Hum Mutat 1999; 13: 99-115. PMID: 10094567
- Oh J, Bailin T, Fukai K, Feng GH, Ho L, Mao J, Frenk E, Tamura N, Spritz RA. Positional cloning of a gene for Hermansky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet 1996:14: 300-6. PMID: 8896559
- Oh J, Ho L, Ala-Mello S, Amato D, Armstrong L, Bellucci S, Carakushansky G, Ellis JP, Fong CT, Green JS, Heon E, Legius E, Levin AV, Nieuwenhuis HK, Pinckers A, Tamura N, Whiteford ML, Yamasaki H, Spritz RA. Mutation analysis of patients with Hermansky-Pudlak syndrome: a frameshift hot spot in the HPS gene and apparent locus heterogeneity. Am J Hum Genet 1998; 62: 593-8. PMID: 9497254
- Sandrock K, Bartsch I, Rombach N, Schmidt K, Nakamura L, Hainmann I, Busse A, Zieger B. Compound heterozygous mutations in 2 siblings with Hermansky-Pudlak syndrome type 1 (HPS1). Klin Padiatr 2010; 222: 168-74. PMID: 20514622
- Shotelersuk V, Hazelwood S, Larson D, Iwata F, Kaiser-Kupfer MI, Kuehl E, Bernardini I, Gahl WA. Three new mutations in a gene causing Hermansky-Pudlak syndrome: clinical correlations. Mol Genet Metab 1998 ; 64: 99-107. PMID: 9705234
- Smith JW, Koshoffer A, Morris RE, Boissy RE. Membranous complexes characteristic of melanocytes derived from patients with Hermansky-Pudlak syndrome type 1 are macroautophagosomal entities of the lysosomal compartment. Pigment Cell Res 2005; 18: 417-26. PMID: 16280007
- Spritz RA, Oh J. HPS gene mutations in Hermansky-Pudlak syndrome. Am J Hum Genet. 1999; 64: 658. (No Abstract)
- Starcevic M, Nazarian R, Dell'Angelica EC. The molecular machinery for the biogenesis of lysosome-related organelles: lessons from Hermansky-Pudlak syndrome. Semin Cell Dev Biol 2002; 13: 271-8. PMID: 12243726
- Suzuki T, Ito S, Inagaki K, Suzuki N, Tomita Y, Yoshino M, Hashimoto T. Investigation on the IVS5 +5G --> a splice site mutation of HPS1 gene found in Japanese patients with Hermansky-Pudlak syndrome. J Dermatol Sci 2004; 36: 106-8. PMID: 15519141
- Suzuki T, Li W, Zhang Q, Karim A, Novak EK, Sviderskaya EV, Hill SP, Bennett DC, Levin AV, Nieuwenhuis HK, Fong CT, Castellan C, Miterski B, Swank RT, Spritz RA. Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet 2002; 30: 321-4. PMID: 11836498
- Vincent LM, Adams D, Hess RA, Ziegler SG, Tsilou E, Golas G, O'Brien KJ, White JG, Huizing M, Gahl WA. Hermansky-Pudlak syndrome type 1 in patients of Indian descent. Mol Genet Metab 2009; 97:227-33. PMID: 19398212
- Wei AH, Li W. Hermansky-Pudlak syndrome: pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell Melanoma Res 2013; 26: 176-92. PMID: 23171219
- Wei A, Lian S, Wang L, Li W. The first case report of a Chinese Hermansky-Pudlak syndrome patient with a novel mutation on HPS1 gene. J Dermatol Sci. 2009; 56: 130-2. PMID: 19665357
- Wei A, Wang Y, Long Y, Wang Y, Guo X, Zhou Z, Zhu W, Liu J, Bian X, Lian S, Li W. A comprehensive analysis reveals mutational spectra and common alleles in chinese patients with oculocutaneous albinism. J Invest Dermatol 2010; 130: 716-24. PMID: 19865097
- Wei A, Yang X, Lian S, Li W. Implementation of an optimized strategy for genetic testing of the Chinese patients with oculocutaneous albinism. J Dermatol Sci 2011; 62: 124-7. PMID: 21458243
- Wildenberg SC, Fryer JP, Gardner JM, Oetting WS, Brilliant MH, King RA. Identification of a novel transcript produced by the gene responsible for the Hermansky-Pudlak syndrome in Puerto Rico. J Invest Dermatol 1998; 110: 777-81. PMID: 9579545
EDIT HISTORY:
Created by Wei Li: 05/24/2004
Updated by Wei Li: 06/25/2005
Updated by Wei Li: 11/23/2005
Updated by Wei Li: 05/08/2009
Updated by Wei Li: 03/15/2010
Updated by Wei Li: 05/25/2011
Updated by Wei Li: 06/28/2012
Updated by Wei Li: 06/13/2013