GENOMIC
Mapping
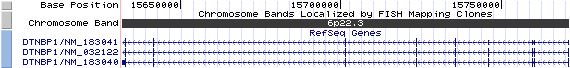
6p22.3. View the map and BAC contig (data from UCSC genome browser).

Structure
(assembly 07/03)
Isoform a (NM_032122): 10 exons, 140,232 bp, chr6:15,631,019-15,771,250.
Isoform b (NM_183040): 9 exons, 139,483 bp, chr6:15,631,768-15,771,250.
Isoform c (NM_183041): 10 exons, 140,232 bp, chr6:15,631,019-15,771,250.
1) NM_183040 contains an additional segment in the coding region compared to NM_032122. The resulting isoform (b) contains a shorter and distinct C-terminus compared to isoform (a).
2) NM_183041 contains an alternate splice site in the 5' coding region and uses a downstream start codon, compared to NM_032122. The encoded isoform (c) has a shorter N-terminus compared to isoform (a).
The figure shows the comparison of these three isoforms (data from UCSC genome browser).
Regulatory Element
Search the 5'UTR and 1kb upstream regions of isoform a (seq1=human HPS7, seq2=mouse Hps7) by CONREAL with 80% Position Weight Matrices (PWMs) threshold (view results here).
TRANSCRIPT
RefSeq/ORF
a) Transcript variant 1 (NM_032122), 1,464 bp, view ORF and the alignment to genomic.
b) Transcript variant 2 (NM_183040), 1,949 bp, view ORF and the alignment to genomic.
c) Transcript variant 3 (NM_183041), 1,509 bp, view ORF and the alignment to genomic.
Expression Pattern
Tissue specificity: ubiquitous. In adult brain, dysbindin mRNA was detected in the frontal cortex, temporal cortex, hippocampus, caudate, putamen, nucleus accumbens, amygdala, thalamus, and midbrain. Overall, dysbindin mRNA is expressed much more prominently in gray matter areas relative to white matter areas. Cortical dysbindin mRNA levels varied statistically significantly according to dysbindin genotype. Variation in dysbindin mRNA levels may be determined in part by variation in the promoter and the 5' and 3' untranslated regions (Weickert, et al (2004)). Bray, et al (2003) also found that the allelic expression differences exceeded 50% in the brain, which suggests that cis-acting variation in gene expression occurs. A defined schizophrenia risk haplotype tags one or more cis-acting variants that results in a relative reduction in DTNBP1 mRNA expression in human cerebral cortex (Bray, et al (2005)). Genome-wide linkage of DTNBP1 expression indicates that a locus on chromosome 8p (putative NRG1) exerts a trans-acting effect on DTNBP1 expression (Bray, et al (2008)).
Immunoelectron microscopy showed that dysbindin-1 is located in (i) synaptic vesicles of axospinous terminals in the dentate gyrus inner molecular layer and CA1 stratum radiatum and in (ii) postsynaptic densities and microtubules of dentate hilus neurons and CA1 pyramidal cells (Telbot, et al (2006)). Tests on the subsynaptic tissue fractions revealed that each isoform is predominantly, if not exclusively, associated with synaptic vesicles (dysbindin-1B) or with postsynaptic densities (dysbindin-1A and -1C). Given the distinctive subsynaptic localization of dysbindin-1A, -1B, and -1C across brain regions, the observed pSTG reductions in dysbindin-1A are postsynaptic and may promote dendritic spine loss with consequent disruption of auditory information processing, while the noted HF reductions in dysbindin-1B and -1C are both presynaptic and postsynaptic and could promote deficits in spatial working memory (Telbot, et al (2011)).
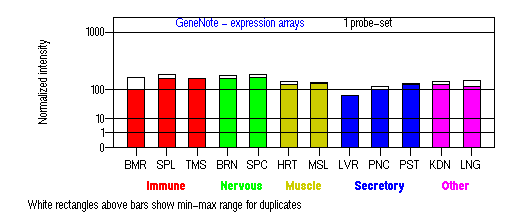
BMR: Bone marrow; SPL: Spleen; TMS: Thymus; BRN: Brain; SPC: Spinal cord; HRT: Heart; MSL: Skeletal muscle; LVR; Liver; PNC: Pancreas; PST: Prostate; KDN: Kidney; LNG: Lung. (data from GeneCards )
PROTEIN
Sequence
Dysbindin-1 (isoform a) (NP_115498): 351aa, ExPaSy NiceProt view of Swiss-Prot: Q96EV8.
Dysbindin-2 (isoform b) ( NP_898861): 303aa.
Dysbindin-3 (isoform c) (NP_898862 ): 270aa.
Synonyms: Hermansky-Pudlak syndrome 7 protein; My031 protein.
Ortholog
| Species | Mouse | Zebrafish | Drosophila |
| GeneView | sdy/Dtnbp1 | 394109 | CG6856 |
| Protein | NP_080048 (352aa) | NP_957428 (362aa) | Q9VVT5 (288aa) |
| Identities | 79%/350aa | 65%/342aa | 28%/219aa |
View multiple sequence alignment (PDF file) by ClustalW and GeneDoc.
Domain
(1) Dysbindin domain: 173-331, CDD: 23538 (Pfam04440).
(2) Domains predicted by SMART(isoform a):
a) coiled coil: 95-180
b) low complexity: 315-329
(3) Transmembrane domains predicted by SOSHI(isoform a): none (predicted as a soluble protein).
(4) Graphic view of InterPro domain structure.
Motif/Site
(1) Predicted results by ScanProsite(isoform a):
a) N-glycosylation site [pattern] [Warning: pattern with a high probability of occurrence]:
117 - 120 NLTH
b) Protein kinase C phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
4 - 6 TlR, 24 - 26 SdK, 168 - 170 TfK, 337 - 339 TdR
c) N-myristoylation site [pattern] [Warning: pattern with a high probability of occurrence]:
226 - 231 GSmsSM, 259 - 264 GgeeNT, 345 - 350 GgedSD
d) Tyrosine sulfation site [rule] [Warning: rule with a high probability of occurrence]:
48 - 62 glellsrYedtwaal
e) cAMP- and cGMP-dependent protein kinase phosphorylation site [pattern] [Warning: pattern with a high probability of occurrence]:
89 - 92 KKkT, 90 - 93 KKtS
f) Leucine zipper pattern [pattern] [Warning: pattern with a high probability of occurrence]:
97 - 118 LqeqlqqLpaliadLesmtanL
(2) Predicted results of subprograms by PSORT II(isoform a):
a) N-terminal signal peptide: none
b) KDEL ER retention motif in the C-terminus: none
c) ER Membrane Retention Signals: none
d) VAC possible vacuolar targeting motif: none
e) Actinin-type actin-binding motif: type 1: none; type 2: none
f) Prenylation motif: none
g) memYQRL transport motif from cell surface to Golgi: none
h) Tyrosines in the tail: none
i) Dileucine motif in the tail: none
3D Model
(1) ModBase: none.
(2) 3D models of isoform (a) predicted by SPARKS (fold recognition) below. View the models by PDB2MGIF.

2D-PAGE
This protein does not exist in the current release of SWISS-2DPAGE.
Computed theoretical MW=39,493Da, pI=4.59 (isoform a).
Computed theoretical MW=34,831Da, pI=5.17 (isoform b).
Computed theoretical MW=30,387Da, pI=4.36 (isoform c).
FUNCTION
Ontology
a) Biological process: visual perception.
b) Plays a role in the biogenesis of lysosome-related organelles such as platelet dense granule and melanosomes.
Location
Mostly cytoplasmic. Also localized to synaptic vesicles and postsynaptic density (PSD). Dysbindin-1 is likely a nucleocytoplasmic shuttling protein and translocated to the nucleus upon treatment with leptomycin B, an inhibitor of exportin-1/CRM1-mediated nuclear export. Dysbindin-1 harbors a functional nuclear export signal necessary for its nuclear export, and the nucleocytoplasmic shuttling of dysbindin-1 affects its regulation of synapsin I expression (Fei, et al).
Interaction
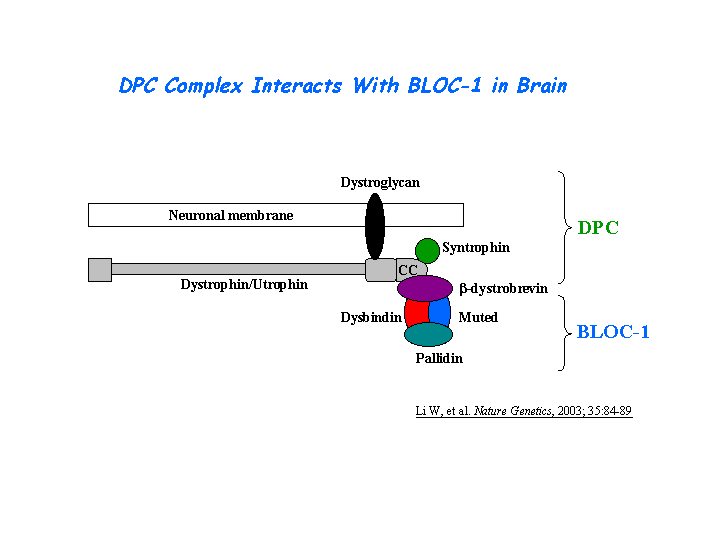
Binds to alpha and beta dystrobrevins that are components of the dystrophin-associated protein complex (DPC) (Benson, et al(2001); Li, et al (2003)) (view diagram of DPC and BLOC-1 interaction here). Nazarian, et al demonstrated direct interaction involving coiled-coil-forming regions in both dysbindin and the dystrobrevins. However, recombinant proteins bearing the coiled-coil-forming regions of the dystrobrevins failed to bind endogenous BLOC-1 from HeLa cells or mouse brain or muscle, suggesting that dysbindin assembled into BLOC-1 is not a physiological binding partner of the dystrobrevins. Benson, et al (2004) showed that dysbindin binds to a novel 413-kDa protein, myospryn, which is expressed in cardiac and skeletal muscle. The C terminus of myospryn contains BBC, FN3, and SPRY domains in a configuration reminiscent of the tripartite motif protein family, as well as the dysbindin-binding site and a region mediating self-association. Desmin binds to myospryn,dysbindin and pallidin. The N-terminal 1-103 fragment of desmin binds to myospryn carboxyl terminus through the SPRY domain (Kouloumenta, et al ). Yeast two-hybrid screening identified the direct interaction of a novel RING finger protein RNF151 and dysbindin, which was confirmed by the co-immunoprecipitation of the proteins and by their co-localization in intact cells, suggesting a role in acrosome formation (Nian, et al ). Yin, et al suggested that the acidic domain of dysbindin and its paralogs in humans may function to recruit casein kinase-1 isoforms to protein complexes involved in multiple biological functions. In the yeast two-hybrid system, TRIM32 binds and ubiquitinates dysbindin (Locke, et al ). A specific interaction between the domains of DISC1 (316-597) and dysbindin (82-173) (Ottis, et al ).
{kind=link}
Dysbindin interacts with pallidin, muted and snapin (Li, et al (2003); Starcevic, et al). Dysbindin is a subunit of the biogenesis of lysosome-related organelles complex 1 (BLOC-1), which is composed of the products of Dtnbp1 and seven other HPS genes, mu, pa, cno, rp, Snapap, Blos1, Blos2 (Ciciotte, et al; Falcon-Perez , et al; Li, et al (2003); Moriyama, et al; Starcevic, et al) (view diagram of BLOC-1 complex here). The BLOC-1 complex appears as a linear chain of eight globular domains, ?300 ? long and ?30 ? in diameter. Two stable subcomplexes were defined, pallidin-Cappuccino-BLOS1 and dysbindin-Snapin-BLOS2. Both subcomplexes are 1:1:1 heterotrimers (Lee, et al). Within BLOC-1, the same 69-residue region of dysbindin that is sufficient for dystrobrevin binding in vitro also contains the binding sites for pallidin and snapin, and at least part of the muted-binding interface (Nazarian, et al). Tissue fractionation of whole mouse brains and human hippocampal formations revealed that both dysbindin-1 and snapin are concentrated in tissue enriched in synaptic vesicle membranes and less commonly in postsynaptic densities. It is not detected in presynaptic tissue fractions lacking synaptic vesicles (Talbot, et al (2006) ).
{kind=link}
BLOC-1 is thought to be involved in endosomal transport. Comparative proteomics of clathrin-coated vesicles (CCVs) have identified seven components of BLOC-1 (Borner, et al). BLOC-1 and AP-3 protein complexes affect the targeting of SNARE and non-SNARE AP-3 cargoes and suggest a function of the BLOC-1 complex in membrane protein sorting (Salazar, et al). BLOC-1 interacts physically and functionally with AP-3 to facilitate the trafficking of a known AP-3 cargo, CD63, and of tyrosinase-related protein 1 (Tyrp1). BLOC-1 also interacts with BLOC-2 to facilitate Tyrp1 trafficking by a mechanism apparently independent of AP-3 function. Both BLOC-1 and -2 localize mainly to early endosome-associated tubules as determined by immunoelectron microscopy (Di Pietro, et al). BLOC-1-deficient melanocytes accumulate the melanosomal protein Tyrp1, but not other melanosomal proteins, in endosomal vacuoles and the cell surface due to failed biosynthetic transit from early endosomes to melanosomes and consequent increased endocytic flux. Melanocytes from HPS model mice lacking a different protein complex, BLOC-2, accumulate Tyrp1 in distinct downstream endosomal intermediates, suggesting that BLOC-1 and BLOC-2 act sequentially in the same pathway. By contrast, intracellular Tyrp1 is correctly targeted to melanosomes in melanocytes lacking another HPS-associated protein complex, AP-3 (Setty, et al). Dysbindin formed a complex with the AP-3 complex through the direct binding to its mu subunit (Taneichi-Kuroda, et al). BLOC-1 interactions with the COG complex, a Golgi apparatus tether, and antioxidant enzymes peroxiredoxins 1-2(Gokhale, et al).
Overexpression of dysbindin induced the expression of two pre-synaptic proteins, SNAP25 and synapsin I, and increased extracellular basal glutamate levels and release of glutamate evoked by high potassium. Conversely, knockdown of endogenous dysbindin protein by small interfering RNA (siRNA) resulted in the reduction of pre-synaptic protein expression and glutamate release, suggesting that dysbindin might influence exocytotic glutamate release via upregulation of the molecules in pre-synaptic machinery (Numakawa, et al). Munc18-1 was co-immunoprecipitated with dysbindin from rat brain lysate, and directly interacted with dysbindin in vitro. In primary cultured rat hippocampal neurons, a part of dysbindin was co-localized with Munc18-1 at pre-synaptic terminals (Hikita, et al). In situ hybridization analysis showed the mRNA expression of dysbindin in the mouse substantia nigra. Suppression of dysbindin expression in PC12 cells resulted in an increase of the expression of SNAP25, and increased the release of dopamine. On the other hand, up-regulation of dysbindin expression in PC12 cells showed a tendency to decrease the expression of SNAP25. These suggest that dysbindin might regulate the dopamine release in dopaminergic cells via modulation of the expression of SNAP25 (Kumamoto, et al). PI4KIIalpha copurified with BLOC-1 and AP-3 in neuronal cells. PI4KIIalpha was targeted to processes in wild-type primary cultured cortical neurons and PC12 cells but failed to reach neurites in cells lacking either AP-3 or BLOC-1 (Larimore, et al).
DTNBP1 siRNA increased cell surface DRD2 and blocked dopamine-induced DRD2 internalization. MUTED siRNA produced similar effects. In contrast, decreased dysbindin did not change dopamine D1 receptor (DRD1) levels, or its basal or dopamine-induced internalization (Iizuka, et al). Dysbindin co-immunoprecipitated with GASP-1 (or GPRASP-1), a cytoplasmic protein shown previously to modulate lysosomal trafficking of D2 dopamine and delta opioid receptors by direct interaction (Marley, et al).
GST pull-down from human neuroblastoma lysates showed an association of dysbindin-1 with the DNA-dependent protein kinase (DNA-PK) complex. The DNA-PK complex interacts only with splice isoforms A and B, but not with C which lacks exons 1-6. Isoforms A and B localized in nucleus, where the kinase complex exist, whereas the isoform C was found exclusively in cytosol. Phosphorylation assay shows that the DNA-PK complex phosphorylated dysbindin-1 isoforms A and B, suggesting that DNA-PK regulates the dysbindin-1 isoforms A and B by phosphorylation in nucleus (Oyama, et al).
In the pathogeneisis of dysbindin-related schizophrenia, neurodevelopmenal defects have been noted. dysbindin-1, WAVE2 and Abi-1 form a ternary complex, and dysbindin-1 promotes the binding of WAVE2 to Abi-1. It is suggested that dysbindin-1 at the postsynapse regulates dendritic spine morphogenesis through the interaction with WAVE2 and Abi-1 (Ito, et al). In addition, necdin is a binding partner of dysbindin-1 by using a yeast two-hybrid screen. Dysbindin-1 recruits necdin to the cytoplasm, thereby attenuating the repressive effects of necdin on p53 transcriptional activity. Knockdown of dysbindin-1, like knockdown of p53, greatly decreases the expressions of the p53 target genes coronin 1b and rab13, which are required for neurite outgrowth (Ma, et al).
Dysbindin and DISC1 share common PPIs suggesting they may affect common biological processes and that the function of schizophrenia risk genes may converge (Camargo, et al). A direct interaction of soluble and insoluble DISC1 protein with dysbindin protein demonstrates convergence of so far considered independent mental disease genes by direct molecular interaction (Ottis, et al). Guo, et al constructed a DTNBP1 interactome. The link between dysbindin and DISC1 is suggested by the interactions with members of the exocyst, dynactin and chaperonin containing T-complex protein complexes (Mead, et al).
Besides BLOC-1 and dystrophin-associated protein complex, several molecules in the DTNBP1 network likely provide insight into the role of DTNBP1 in biological systems: retinoic acid, beta-estradiol, calmodulin and tumour necrosis factor.
HPS7p drosophila homolog CG6856 interaction information in CuraGen interaction database.
Pathway
HPS7 may play a role in organelle biogenesis associated with melanosomes, platelet dense granules, and lysosomes (view diagram of BLOC-1 pathway here). Melanosome maturation requires at least two cargo transport pathways directly from early endosomes to melanosomes, one mediated by AP-3 and another by BLOC-1 and BLOC-2 (Setty, et al).
{kind=link}
MUTATION
Allele or SNP
1 allelic variant described in OMIM.
SNPs deposited in dbSNP.
Distribution
| Location | Genomic | cDNA | Protein | Type | Ethnicity | Reference |
| Exon 4 | 177G>A | 177G>A | W59X | nonsense | Caucasian | Lowe, et al (2013) |
| Exon 5 | 307C>T | 307C>T | Q103X | nonsense | Portuguese | Li, et al (2003) |
Effect
The Q103X mutation (Li, et al (2003)) is predicted to cause nonsense mediated decay (NMD).
PHENOTYPE
Mutation in the HPS7 gene causes Hermansky-Pudlak syndrome type 7 (HPS-7, OMIM 607145) (Li, et al (2003)). A 77 years old women with a homozygous HPS7 nonsense mutation (W59X) exhibits typical HPS symtoms together with colitis, but without pulmonary dysfunction (Lowe, et al).
A significant association between schizophrenia 3 (SCZD3, OMIM 600511) and various dysbindin-1 haplotypes has been reported in populations from England, Ireland (O'Donovan, et al; Straub, et al; van den Oord, et al; Williams, et al), Wales (Williams, et al), The Netherlands, Germany/Hungary/Israel (Schwab, et al), Sweden (Van Den Bogaert, et al), Bulgaria (Kirov, et al), China (Tang, et al (2003); Li, et al (2005)), Japan (Numakawa, et al), USA (Funke, et al). Assocoation studies have also shown that risk DTNBP1 haplotypes may influence human intelligence or neurocognitive dysfunction (bipolar disorder, negative symptoms of schizophrenia) (Burdick, et al (2006); Derosse, et al; Fallin, et al). The DTNBP1 SNP rs2619522 had significant effects bilaterally in the hippocampus as well as in the anterior middle frontal gyrus and the intraparietal cortex. Carriers of the G allele showed significantly higher grey matter volumes in these brain regions than T/T homozygotes (Trost, et al). Carriers of the CTCTAC haplotype, demonstrated a significantly greater decline in IQ as compared with non-carriers (p=0.05), suggesting that DTNBP1 influences the severity of intellectual decline in schizophrenia (Burdick, et al (2007)). However, some studies have shown no association or inconsistency between DTNBP1 and SCZD (Mutsuddi, et al; Wood, et al; Turunen, et al; Liu, et al; Sanders, et al). An epistatic interaction between DTNBP1 and MUTED contributing to SZ in the absence of a significant main effect at MUTED was reported (Morris, et al).
Talbot, et al (2004) found that 73-93% of cases in two schizophrenia populations displayed presynaptic dysbindin-1 reductions averaging 18-42% (P = 0.027-0.0001) at hippocampal formation sites lacking neuronal dystrobrevin (i.e., beta-dystrobrevin). An inversely correlated increase in vesicular glutamate transporter-1 (VGluT-1) occurred in DGiml of the same schizophrenia cases. Weickert, et al (2004) also found that patients with schizophrenia had statistically significantly reduced (15% to 20%) dysbindin mRNA levels in multiple layers of the dorsolateral prefrontal cortex and possibly in the midbrain. Dysbindin-1 mRNA in the hippocampal formation of patients with schizophrenia was decreased and the reduced expression was found in dentate granule and polymorph cells and in hippocampal field CA3, but not in CA1 (Weickert, et al (2008)) . RNA expression of DTNBP1 and NRG1 in immortalized lymphocytes of SZ patients was lower than in controls before and after olanzapine stimulation (Chagnon, et al ).
In dysbindin-null sdy mice, loss of dysbindin does not have obvious changes of the histological hippocampal structures (Feng, et al). However, ultrastructural abnormalities were observed in sdy hippocampal synapses. These include larger synaptic vesicle size, lower vesicle number, shorter active zone, narrower synaptic cleft, and thicker PSD (Chen, et al). These changes may alter the synaptic transmission efficacy, which results in the schizophria-like symptoms in sdy mice, such as impaired social interaction and recognition/memory (Feng, et al). Furthermore, the steady-state level of snapin is reduced to 75% of the level in wild-type mice (Feng, et al). The kinetics of synaptic exocytosis is impaired in a similar pattern in both sdy and snapin knockout mice (Chen, et al; Tian et al). Disruption of the dysbindin gene dramatically increased NR2A-mediated synaptic currents, without affecting AMPA receptor currents, in hippocampal CA1 neurons. The Dys-/- hippocampal slices exhibited an enhanced LTP, whereas basal synaptic transmission, presynaptic properties, and LTD were normal (Tang, et al (2009) ). In addition, disruption of dysbindin gene resulted in a marked decrease in the excitability of fast-spiking (FS) GABAergic interneurons in both PFC and striatum (Ji, et al ). Similarly, in the drosophila, dysbindin is required presynaptically for the retrograde, homeostatic modulation of neurotransmission, and functions in a dose-dependent manner downstream or independently of calcium influx (Dickman, et al ). Taken together, these provide a possible mechanism that lower expression of dysbindin may contribute to the pathogenesis of schizophrenia.
Interestingly, dysbindin-1 is up-regulated in the dystrophin-deficient muscle of a mouse model of Duchenne muscular dystrophy, the mdx mutant (Benson, et al (2001)). Moreover, Sillitoe, et al reported that there is a dramatic increase of the number of dysbindin-immunoreactive glomeruli in the posterior cerebellar vermis in the mdx mutant. Abnormal synaptic organization in the cerebellum may contribute to the neurological problems associated with muscular dystrophy and schizophrenia.
However, thus far, no deleterious mutations of the DTNBP1 gene have been identified in any of the schizophrenic patients of these studies. Likewise, no apparent schizophrenic symptoms were observed in the HPS-7 patient, null for the DTNBP1 gene (Li, et al (2003); Lowe, et al (2013)).
REFERENCE
- Benson MA, Newey SE, Martin-Rendon E, Hawkes R, Blake DJ. Dysbindin, a novel coiled-coil-containing protein that interacts with the dystrobrevins in muscle and brain. J Biol Chem 2001; 276: 24232-41. PMID: 11316798
- Benson MA, Tinsley CL, Blake DJ. Myospryn is a novel binding partner for dysbindin in muscle. J Biol Chem 2004; 279: 10450-8. PMID: 14688250
- Borner GH, Harbour M, Hester S, Lilley KS, Robinson MS. Comparative proteomics of clathrin-coated vesicles. J Cell Biol 2006; 175: 571-8. PMID: 17116749
- Bray NJ, Buckland PR, Owen MJ, O'Donovan MC. Cis-acting variation in the expression of a high proportion of genes in human brain. Hum Genet 2003; 113: 149-53. PMID: 12728311
- Bray NJ, Preece A, Williams NM, Moskvina V, Buckland PR, Owen MJ, O'Donovan MC. Haplotypes at the dystrobrevin binding protein 1 (DTNBP1) gene locus mediate risk for schizophrenia through reduced DTNBP1 expression. Hum Mol Genet 2005; 14:1947-54. PMID: 15917270
- Bray NJ, Holmans PA, van den Bree MB, Jones L, Elliston LA, Hughes G, Richards AL, Williams NM, Craddock N, Owen MJ, O'Donovan MC. Cis- and Trans- Loci Influence Expression of the Schizophrenia Susceptibility Gene DTNBP1. Hum Mol Genet 2008; 17: 1169-74. PMID: 18182443
- Burdick KE, Goldberg TE, Funke B, Bates JA, Lencz T, Kucherlapati R, Malhotra AK. DTNBP1 genotype influences cognitive decline in schizophrenia.Schizophr Res 2007; 89: 169-72.PMID: 17074466
- Burdick KE, Lencz T, Funke B, Finn CT, Szeszko PR, Kane JM, Kucherlapati R, Malhotra AK. Genetic variation in DTNBP1 influences general cognitive ability. Hum Mol Genet 2006; 15: 1563-8.PMID: 16415041
- Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, Bonnert TP, Whiting PJ, Brandon NJ. Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry 2007; 12: 74-86. PMID: 17043677
- Chagnon YC, Roy MA, Bureau A, M¨¦rette C, Maziade M. Differential RNA expression between schizophrenic patients and controls of the dystrobrevin binding protein 1 and neuregulin 1 genes in immortalized lymphocytes.Schizophr Res 2008; 100: 281-90. PMID: 18234478
- Chen XW, Feng YQ, Hao CJ, Guo XL, He X, Zhou ZY, Guo N, Huang HP, Xiong W, Zheng H, Zuo PL, Zhang X, Li W, Zhou Z. DTNBP1, a schizophrenia susceptiblility gene, affects kinetics of transmitter release. J Cell Biol 2008; 18: 791-801 PMID: 18504299
- Ciciotte SL, Gwynn B, Moriyama K, Huizing M, Gahl WA, Bonifacino JS, Peters LL. Cappuccino, a mouse model of Hermansky-Pudlak syndrome, encodes a novel protein that is part of the pallidin-muted complex (BLOC-1). Blood 2003; 101: 4402-7. PMID: 12576321
- Derosse P, Funke B, Burdick KE, Lencz T, Ekholm JM, Kane JM, Kucherlapati R, Malhotra AK. Dysbindin genotype and negative symptoms in schizophrenia. Am J Psychiatry 2006; 163: 532-4. PMID: 16513878
- Dickman DK, Davis GW. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science 2009; 326: 1127-30. PMID: 19965435
- Di Pietro SM, Falcon-Perez JM, Tenza D, Setty SR, Marks MS, Raposo G, Dell'Angelica EC. BLOC-1 interacts with BLOC-2 and the AP-3 complex to facilitate protein trafficking on endosomes. Mol Biol Cell 2006; 17: 4027-38. PMID: 16837549
- Falcon-Perez JM, Starcevic M, Gautam R, Dell'Angelica EC. BLOC-1, a novel complex containing the pallidin and muted proteins involved in the biogenesis of melanosomes and platelet-dense granules. J Biol Chem 2002; 277: 28191-9. PMID: 12019270
- Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, McGrath JA, Steel G, Nestadt G, Liang KY, Huganir RL, Valle D, Pulver AE. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am J Hum Genet 2005; 77: 918-36. PMID: 16380905
- Fei E, Ma X, Zhu C, Xue T, Yan J, Xu Y, Zhou J, Wang G. Nucleocytoplasmic shuttling of dysbindin-1, a schizophrenia-related protein, regulates synapsin I expression. J Biol Chem 2010; 285: 38630-40. PMID: 20921223
- Feng YQ, Zhou ZY, He X, Wang H,Guo XL, Hao CJ, Guo Y, Zhen XC, Li W. Dysbindin deficiency in sandy mice causes reduction of snapin and displays behaviors related to schizophrenia. Schizophr Res 2008; 106: 218-28. PMID: 18774265
- Funke B, Finn CT, Plocik AM, Lake S, DeRosse P, Kane JM, Kucherlapati R, Malhotra AK. Association of the DTNBP1 locus with schizophrenia in a U.S. population. Am J Hum Genet 2004; 75: 891-8. PMID: 15362017
- Gokhale A, Larimore J, Werner E, So L, Moreno-De-Luca A, Lese-Martin C, Lupashin VV, Smith Y, Faundez V. Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J Neurosci 2012; 32: 3697-711. PMID: 22423091
- Guo AY, Sun J, Riley BP, Thiselton DL, Kendler KS, Zhao Z. The dystrobrevin-binding protein 1 gene: features and networks. Mol Psychiatry 2009; 14: 18-29. PMID: 18663367
- Hikita T, Taya S, Fujino Y, Taneichi-Kuroda S, Ohta K, Tsuboi D, Shinoda T, Kuroda K, Funahashi Y, Uraguchi-Asaki J, Hashimoto R, Kaibuchi K. Proteomic analysis reveals novel binding partners of dysbindin, a schizophrenia-related protein. J Neurochem 2009; 110: 1567-74. PMID: 19573021
- Iizuka Y, Sei Y, Weinberger DR, Straub RE. Evidence that the BLOC-1 protein dysbindin modulates dopamine D2 receptor internalization and signaling but not D1 internalization. J Neurosci 2007; 27: 12390-5. PMID: 17989303
- Ito H, Morishita R, Shinoda T, Iwamoto I, Sudo K, Okamoto K, Nagata K. Dysbindin-1, WAVE2 and Abi-1 form a complex that regulates dendritic spine formation. Mol Psychiatry 2010; 15: 976-86. PMID: 20531346
- Ji Y, Yang F, Papaleo F, Wang HX, Gao WJ, Weinberger DR, Lu B. Role of dysbindin in dopamine receptor trafficking and cortical GABA function. Proc Natl Acad Sci U S A. 2009; 106: 19593-8. PMID: 19887632
- Kirov G, Ivanov D, Williams NM, Preece A, Nikolov I, Milev R, Koleva S, Dimitrova A, Toncheva D, O'Donovan MC, Owen MJ. Strong evidence for association between the dystrobrevin binding protein 1 gene (DTNBP1) and schizophrenia in 488 parent-offspring trios from Bulgaria. Biol Psychiatry. 2004; 55: 971-5. PMID: 15121479
- Kouloumenta A, Mavroidis M, Capetanaki Y. Proper perinuclear localization of the TRIM-like protein myospryn requires its binding partner desmin. J Biol Chem 2007; 282: 35211-21.PMID: 17872945
- Kumamoto N, Matsuzaki S, Inoue K, Hattori T, Shimizu S, Hashimoto R, Yamatodani A, Katayama T, Tohyama M. Hyperactivation of midbrain dopaminergic system in schizophrenia could be attributed to the down-regulation of dysbindin. Biochem Biophys Res Commun 2006; 345: 904-9.PMID: 16701550
- Larimore J, Tornieri K, Ryder PV, Gokhale A, Zlatic SA, Craige B, Lee JD, Talbot K, Pare JF, Smith Y, Faundez V. The schizophrenia susceptibility factor dysbindin and its associated complex sort cargoes from cell bodies to the synapse. Mol Biol Cell 2011; 22: 4854-67. PMID: 21998198
- Lee HH, Nemecek D, Schindler C, Smith WJ, Ghirlando R, Steven AC, Bonifacino JS, Hurley JH. Assembly and architecture of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J Biol Chem 2012; 287: 5882-90. PMID: 22203680
- Li T, Zhang F, Liu X, Sun X, Sham PC, Crombie C, Ma X, Wang Q, Meng H, Deng W, Yates P, Hu X, Walker N, Murray RM, St Clair D, Collier DA. Identifying potential risk haplotypes for schizophrenia at the DTNBP1 locus in Han Chinese and Scottish populations. Mol Psychiatry 2005; 10:1037-44. PMID: 16044171
- Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O'Brien EP, Tinsley CL, Blake DJ, Spritz RA, Copeland NG, Jenkins NA, Amato D, Roe BA, Starcevic M, Dell'Angelica EC, Elliott RW, Mishra V, Kingsmore SF, Paylor RE, Swank RT. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet 2003; 35: 84-9. PMID: 12923531
- Liu CM, Liu YL, Fann CS, Yang WC, Wu JY, Hung SI, Chen WJ, Chueh CM, Liu WM, Liu CC, Hsieh MH, Hwang TJ, Faraone SV, Tsuang MT, Hwu HG. No association evidence between schizophrenia and dystrobrevin-binding protein 1 (DTNBP1) in Taiwanese families. Schizophr Res 2007; 93: 391-8.PMID: 17407805
- Locke M, Tinsley CL, Benson MA, Blake DJ. TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum Mol Genet 2009; 18: 2344-58. PMID: 19349376
- Lowe GC, Sanchez Guiu I, Chapman O, Rivera J, Lordkipanidze M, Dovlatova N, Wilde J, Watson SP, Morgan NV; UK GAPP collaborative. Microsatellite markers as a rapid approach for autozygosity mapping in Hermansky-Pudlak syndrome: identification of the second HPS7 mutation in a patient presenting late in life. Thromb Haemost 2013; 109: 766-8. PMID: 23364359
- Ma X, Fei E, Fu C, Ren H, Wang G. Dysbindin-1, a schizophrenia-related protein, facilitates neurite outgrowth by promoting the transcriptional activity of p53. Mol Psychiatry. 2011; 16: 1105-16.PMID: 21502952
- Marley A, von Zastrow M. Dysbindin promotes the post-endocytic sorting of G protein-coupled receptors to lysosomes. PLoS One 2010; 5:e9325. PMID: 20174469
- Mead CL, Kuzyk MA, Moradian A, Wilson GM, Holt RA, Morin GB. Cytosolic protein interactions of the schizophrenia susceptibility gene dysbindin. J Neurochem. 2010; 113: 1491-503. PMID: 20236384
- Moriyama K, Bonifacino JS. Pallidin is a component of a multi-protein complex involved in the biogenesis of lysosome-related organelles. Traffic 2002; 3: 666-77. PMID: 12191018
- Morris DW, Murphy K, Kenny N, Purcell SM, McGhee KA, Schwaiger S, Nangle JM, Donohoe G, Clarke S, Scully P, Quinn J, Meagher D, Baldwin P, Crumlish N, O'Callaghan E, Waddington JL, Gill M, Corvin AP. Dysbindin (DTNBP1) and the biogenesis of lysosome-related organelles complex 1 (BLOC-1): main and epistatic gene effects are potential contributors to schizophrenia susceptibility. Biol Psychiatry 2008; 63: 24-31. PMID: 17618940
- Mutsuddi M, Morris DW, Waggoner SG, Daly MJ, Scolnick EM, Sklar P. Analysis of high-resolution HapMap of DTNBP1 (Dysbindin) suggests no consistency between reported common variant associations and schizophrenia. Am J Hum Genet 2006; 79: 903-9. PMID: 17033966
- Nazarian R, Starcevic M, Spencer MJ, Dell'Angelica EC. Reinvestigation of the dysbindin subunit of BLOC-1 (biogenesis of lysosome-related organelles complex-1) as a dystrobrevin-binding protein. Biochem J 2006; 395: 587-98. PMID: 16448387
- Nian H, Fan C, Liao S, Shi Y, Zhang K, Liu Y, Han C. RNF151, a testis-specific RING finger protein, interacts with dysbindin. Arch Biochem Biophys 2007; 465: 157-63. PMID: 17577571
- Numakawa T, Yagasaki Y, Ishimoto T, Okada T, Suzuki T, Iwata N, Ozaki N, Taguchi T, Tatsumi M, Kamijima K, Straub RE, Weinberger DR, Kunugi H, Hashimoto R.Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet. 2004;13: 2699-708. PMID: 15345706
- O'Donovan MC, Williams NM, Owen MJ. Recent advances in the genetics of schizophrenia. Hum Mol Genet 2003; 12: R125-33. PMID: 12952866
- Ottis P, Bader V, Trossbach SV, Kretzschmar H, Michel M, Leliveld SR, Korth C. Convergence of two independent mental disease genes on the protein level: recruitment of dysbindin to cell-invasive disrupted-in-schizophrenia 1 aggresomes. Biol Psychiatry 2011; 70: 604-10. PMID: 21531389
- Oyama S, Yamakawa H, Sasagawa N, Hosoi Y, Futai E, Ishiura S. Dysbindin-1, a schizophrenia-related protein, functionally interacts with the DNA- dependent protein kinase complex in an isoform-dependent manner. PLoS ONE 2009; 4: e4199. PMID: 19142223
- Salazar G, Craige B, Styers ML, Newell-Litwa KA, Doucette MM, Wainer BH, Falcon-Perez JM, Dell'Angelica EC, Peden AA, Werner E, Faundez V. BLOC-1 complex deficiency alters the targeting of adaptor protein complex-3 cargoes. Mol Biol Cell 2006; 17: 4014-26. PMID: 16760431
- Sanders AR, Duan J, Levinson DF, Shi J, He D, Hou C, Burrell GJ, Rice JP, Nertney DA, Olincy A, Rozic P, Vinogradov S, Buccola NG, Mowry BJ, Freedman R, Amin F, Black DW, Silverman JM, Byerley WF, Crowe RR, Cloninger CR, Martinez M, Gejman PV. No Significant Association of 14 Candidate Genes With Schizophrenia in a Large European Ancestry Sample: Implications for Psychiatric Genetics. Am J Psychiatry 2008; 165: 497-506. PMID: 18198266
- Schwab SG, Knapp M, Mondabon S, Hallmayer J, Borrmann-Hassenbach M, Albus M, Lerer B, Rietschel M, Trixler M, Maier W, Wildenauer DB. Support for association of schizophrenia with genetic variation in the 6p22.3 gene, dysbindin, in sib-pair families with linkage and in an additional sample of triad families. Am J Hum Genet 2003; 72: 185-90. PMID: 12474144
- Setty SR, Tenza D, Truschel ST, Chou E, Sviderskaya EV, Theos AC, Lamoreux ML, Di Pietro SM, Starcevic M, Bennett DC, Dell'angelica EC, Raposo G, Marks MS. BLOC-1 Is Required for Cargo-specific Sorting from Vacuolar Early Endosomes toward Lysosome-related Organelles. Mol Biol Cell 2007; 18: 768-80. PMID: 17182842
- Sillitoe RV, Benson MA, Blake DJ, Hawkes R. Abnormal dysbindin expression in cerebellar mossy fiber synapses in the mdx mouse model of Duchenne muscular dystrophy. J Neurosci 2003; 23: 6576-85. PMID: 12878699
- Starcevic M, Dell'Angelica EC. Identification of snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J Biol Chem 2004; 279: 28393-401. PMID: 15102850
- Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, Cesare AJ, Gibberman A, Wang X, O'Neill FA, Walsh D, Kendler KS. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet 2002; 71: 337-48. PMID: 12098102
- Talbot K, Cho DS, Ong WY, Benson MA, Han LY, Kazi HA, Kamins J, Hahn CG, Blake DJ, Arnold SE. Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Hum Mol Genet 2006; 15: 3041-54. PMID: 16980328
- Talbot K, Eidem WL, Tinsley CL, Benson MA, Thompson EW, Smith RJ, Hahn CG, Siegel SJ, Trojanowski JQ, Gur RE, Blake DJ, Arnold SE. Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J Clin Invest 2004; 113: 1353-63. PMID: 15124027
- Talbot K, Louneva N, Cohen JW, Kazi H, Blake DJ, Arnold SE. Synaptic dysbindin-1 reductions in schizophrenia occur in an isoform-specific manner indicating their subsynaptic location. PLoS One 2011; 6: e16886. PMID: 21390302
- Tang JX, Zhou J, Fan JB, Li XW, Shi YY, Gu NF, Feng GY, Xing YL, Shi JG, He L. Family-based association study of DTNBP1 in 6p22.3 and schizophrenia. Mol Psychiatry 2003; 8: 1008. (No Abstract)
- Tang TT, Yang F, Chen BS, Lu Y, Ji Y, Roche KW, Lu B. Dysbindin regulates hippocampal LTP by controlling NMDA receptor surface expression. Proc Natl Acad Sci U S A. 2009; 106: 21395-400. PMID: 19955431
- Taneichi-Kuroda S, Taya S, Hikita T, Fujino Y, Kaibuchi K. Direct interaction of Dysbindin with the AP-3 complex via its mu subunit. Neurochem Int 2009; 54: 431-8. PMID: 19428785
- Tian JH, Wu ZX, Unzicker M, Lu L, Cai Q, Li C, Schirra C, Matti U, Stevens D, Deng C, Rettig J, Sheng ZH. The role of Snapin in neurosecretion: snapin knock-out mice exhibit impaired calcium-dependent exocytosis of large dense-core vesicles in chromaffin cells. J Neurosci 2005; 25: 10546-55. PMID: 16280592
- Trost S, Platz B, Usher J, Scherk H, Wobrock T, Ekawardhani S, Meyer J, Reith W, Falkai P, Gruber O. The DTNBP1 (dysbindin-1) gene variant rs2619522 is associated with variation of hippocampal and prefrontal grey matter volumes in humans. Eur Arch Psychiatry Clin Neurosci 2013; 263: 53-63. PMID: 22580710
- Turunen JA, Peltonen JO, Pietil?inen OP, Hennah W, Loukola A, Paunio T, Silander K, Ekelund J, Varilo T, Partonen T, L?nnqvist J, Peltonen L. The role of DTNBP1, NRG1, and AKT1 in the genetics of schizophrenia in Finland. Schizophr Res 2007; 91: 27-36. PMID: 17300918
- Van Den Bogaert A, Schumacher J, Schulze TG, Otte AC, Ohlraun S, Kovalenko S, Becker T, Freudenberg J, Jonsson EG, Mattila-Evenden M, Sedvall GC, Czerski PM, Kapelski P, Hauser J, Maier W, Rietschel M, Propping P, Nothen MM, Cichon S. The DTNBP1 (dysbindin) gene contributes to schizophrenia, depending on family history of the disease. Am J Hum Genet 2003; 73: 1438-43. PMID: 14618545
- van den Oord EJ, Sullivan PF, Jiang Y, Walsh D, O'Neill FA, Kendler KS, Riley BP. Identification of a high-risk haplotype for the dystrobrevin binding protein 1 (DTNBP1) gene in the Irish study of high-density schizophrenia families. Mol Psychiatry 2003; 8: 499-510. PMID: 12808430
- Weickert CS, Straub RE, McClintock BW, Matsumoto M, Hashimoto R, Hyde TM, Herman MM, Weinberger DR, Kleinman JE. Human Dysbindin (DTNBP1) Gene Expression in Normal Brain and in Schizophrenic Prefrontal Cortex and Midbrain. Arch Gen Psychiatry. 2004; 61: 544-55. PMID: 15184234
- Weickert CS, Rothmond DA, Hyde TM, Kleinman JE, Straub RE. Reduced DTNBP1 (dysbindin-1) mRNA in the hippocampal formation of schizophrenia patients.Schizophr Res 2008; 98: 105-10. PMID: 17961984
- Williams NM, Preece A, Morris DW, Spurlock G, Bray NJ, Stephens M, Norton N, Williams H, Clement M, Dwyer S, Curran C, Wilkinson J, Moskvina V, Waddington JL, Gill M, Corvin AP, Zammit S, Kirov G, Owen MJ, O'Donovan MC. Identification in 2 independent samples of a novel schizophrenia risk haplotype of the dystrobrevin binding protein gene (DTNBP1). Arch Gen Psychiatry 2004 ; 61: 336-44. PMID: 15066891
- Wood LS, Pickering EH, Dechairo BM. Significant support for DAO as a schizophrenia susceptibility Locus: examination of five genes putatively associated with schizophrenia. Biol Psychiatry 2007; 61: 1195-9. PMID: 17055463
- Yin H, Laguna KA, Li G, Kuret J. Dysbindin structural homologue CK1BP is an isoform-selective binding partner of human casein kinase-1. Biochemistry 2006; 45: 5297-308. PMID: 16618118
EDIT HISTORY:
Created by Wei Li: 06/02/2004
Updated by Wei Li: 06/01/2005
Updated by Wei Li: 04/06/2006
Updated by Wei Li: 12/26/2006
Updated by Wei Li: 02/28/2006
Updated by Wei Li: 08/20/2008
Updated by Wei Li: 02/24/2009
Updated by Wei Li: 05/24/2011
Updated by Wei Li: 08/02/2012
Updated by Wei Li: 06/13/2013